Registo de Dispositivos Médicos US FDA - Visão Geral
Os Estados Unidos da América (EUA) são conhecidos por serem um mercado altamente regulamentado para Dispositivos Médicos, com percursos e requisitos de registo bem definidos. Os regulamentos iniciais para dispositivos médicos nos US datam de 1976 e evoluíram ao longo do tempo. São regulamentados pelo Centro de Dispositivos e Saúde Radiológica (CDRH) sob a Food and Drug Administration (FDA). A Freyr ajudou vários fabricantes de dispositivos a cumprir o processo de registo de dispositivos médicos da US FDA.
![]()
Autoridade Regulamentar: Food and Drug Administration (FDA)![]()
Regulamento: Título 21 do Código de Regulamentos Federais (21 CFR) Partes 800 – 1299![]()
Via Regulamentar: Notificação Pré-Comercialização ou Aprovação pré-comercialização ou Classificação De Novo![]()
Representante Autorizado: Agente US![]()
Requisito de SGQ: Regulamento do Sistema de Qualidade (QSR) (21 CFR parte 820)![]()
Avaliação de Dados Técnicos: Centro de Dispositivos e Saúde Radiológica![]()
Validade da Licença: Ilimitada![]()
Requisitos de Rotulagem: 21 CFR Parte 801![]()
Formato de Submissão: Papel e CD/DVD![]()
Idioma: Inglês
Classificação de Dispositivos Médicos US
A FDA classifica os Dispositivos Médicos em 3 categorias baseadas no risco: Classe I, Classe II e Classe III, sendo que os dispositivos de Classe I são considerados de baixo risco e os de Classe III estão associados a alto risco. Os requisitos de registo e o percurso variam consoante a classe do dispositivo.
| Classe de Dispositivo | Risco | Via de Registo para aprovação |
|---|---|---|
| I | Baixo Risco | Isento de 510(k) |
| II | Risco Moderado (Com dispositivo de referência) | Notificação Pré-Comercialização/510(k) |
Risco Moderado (Sem dispositivo de referência) | Submissão De-Novo | |
| III | Alto Risco | Aprovação Pré-Comercialização (PMA) |
Agente da US FDA
Empresas sem escritórios locais nos US devem nomear um Agente da FDA dos US para representar o fabricante. O agente da FDA dos US deve residir nos US ou manter um local de negócios nos US. As responsabilidades a serem cumpridas pelo agente são predeterminadas pela FDA dos US como parte dos regulamentos CFR.
Navegue pelas Perguntas Frequentes (FAQs) sobre o Agente US.
Reuniões Interativas com a USFDA
A US FDA apoia os fabricantes através de vários tipos de reuniões de Q-Submission para cumprir diferentes objetivos. Tais reuniões com a agência, antes do início ou durante o desenvolvimento do dispositivo, e antes da submissão de submissões de registo de dispositivos médicos da US FDA, ajudam os fabricantes a otimizar os prazos e os custos incorridos para a comercialização do dispositivo.
Registo de Dispositivos Médicos US
Os dispositivos podem ser aprovados pelo CDRH, FDA, através de qualquer um dos vários percursos de registo. São listados como:
Dispositivos Médicos de Classe I: Os dispositivos de classe I são geralmente isentos de GMP e de submissão 510(k) e não requerem aprovação prévia da US FDA para serem comercializados nos US. Outros requisitos, como o registo do estabelecimento, a listagem de dispositivos, UDI, PMS, etc., devem ser cumpridos pelo fabricante.
Dispositivos médicos de Classe II: Dispositivos de risco médio com dispositivos de referência aprovados via 510(k) podem optar pela Notificação Pré-mercado 510(k) (PMN), também conhecida como registo 510(k). O dispositivo em questão deve estabelecer Equivalência Substancial (SE) com os dispositivos de referência identificados e reivindicados. Esta via é a mais amplamente adotada para o registo de dispositivos nos US. Os fabricantes de dispositivos de risco médio sem dispositivos de referência podem solicitar a classificação pela US FDA através de submissões De-Novo.
Dispositivos Médicos de Classe III: Os fabricantes de dispositivos de Classe III de alto risco devem submeter uma submissão de Aprovação Pré-mercado (PMA) à US FDA. Os dispositivos devem ser submetidos a uma avaliação clínica detalhada e o fabricante deve submeter dados detalhados de segurança e eficácia de estudos clínicos. A US FDA realizará uma inspeção QMS como parte da avaliação antes de emitir uma Aprovação Pré-mercado para o dispositivo.
Registos de Dispositivos Médicos Não-CDRH
Com base nas indicações de uso, alguns produtos de fronteira considerados dispositivos médicos noutros países, como respiradores cirúrgicos, desinfetantes e produtos combinados, envolvem outras Agências, como o Centre for Disease Control (CDC), o National Institute for Occupational Safety and Hazards (NIOSH), a Environmental Protection Agency (EPA), o Centre for Biological Evaluation and Research (CBER) e o Centre for Drug Evaluation and Research (CDER).
Requisitos de Conformidade Pós-Aprovação para Dispositivos Médicos
Todos os fabricantes de dispositivos devem cumprir os requisitos de pós-aprovação listados abaixo:
- Requisito de Registo e Listagem: Os estabelecimentos de todas as classes de dispositivos devem ser registados na base de dados FURLs e o dispositivo deve ser listado após a obtenção da aprovação e antes da comercialização do dispositivo nos US. Alguns dispositivos, como os dispositivos de radiação, devem cumprir outros requisitos, como o número de acesso, antes de poderem ser importados para os US.
- Identificação Única de Dispositivo: Todas as classes de dispositivos devem cumprir as regulamentações de Identificação Única de Dispositivo (UDI) para comercializar os dispositivos nos US.
- Taxas de Estabelecimento: O fabricante deve pagar as taxas anuais de estabelecimento para manter o seu registo de estabelecimento ativo e continuar a comercializar dispositivos nos US. A US FDA tem uma estrutura de taxas reduzida para entidades mais pequenas com Certificado de Pequena Empresa ativo.
- Auditorias de Qualidade: Para dispositivos que não estão isentos de BPF, a US FDA pode inspecionar o estabelecimento de fabrico a qualquer momento para verificar a conformidade com os Regulamentos de Sistemas de Qualidade (QSR), de acordo com o 21 CFR 820.
Fluxo do processo
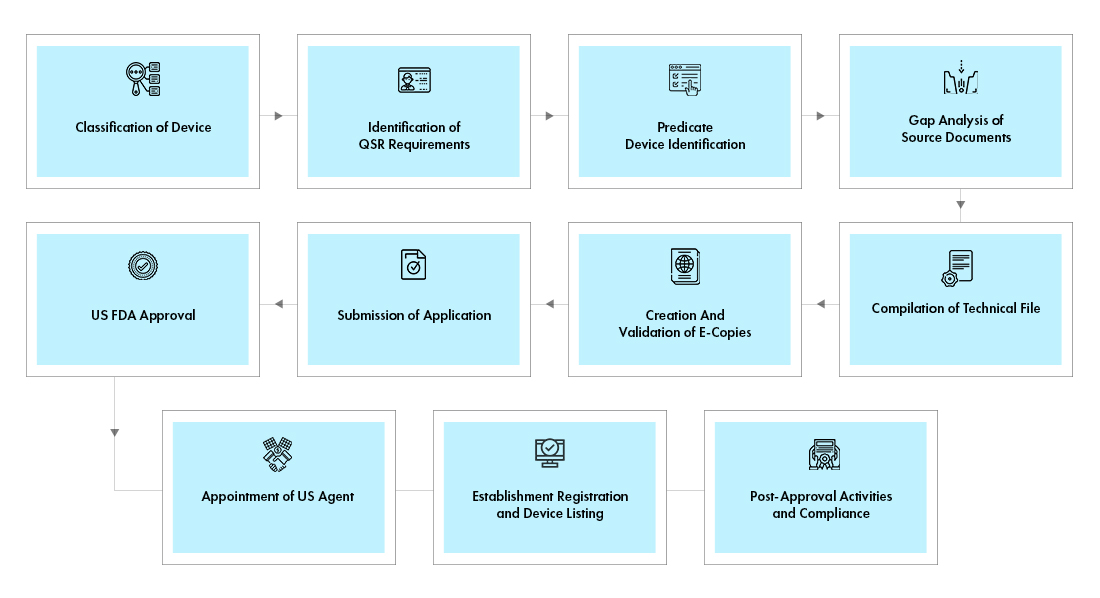
Gestão do Ciclo de Vida do Dispositivo Pós-Aprovação
A Freyr apoia fabricantes estrangeiros na gestão End-to-End do ciclo de vida de dispositivos médicos, incluindo atividades pós-aprovação, tais como:
- Gestão de alterações pós-aprovação - modificações às aprovações de dispositivos médicos existentes, tais como, adição de novas variantes, acessórios; adição de novas indicações de utilização, entre outras
- Manutenção de aprovações e registos através do pagamento atempado das taxas MDUFA à FDA.
- Interligação entre a US FDA e o Fabricante
A Freyr possui um centro de entrega exclusivo nos EUA com uma equipa profissional para fornecer apoio regulatório aos fabricantes na manutenção da qualidade e segurança necessárias para a aprovação. Os especialistas em inteligência da Freyr observam atentamente as atualizações regulatórias e mantêm os clientes informados sobre os passos a serem dados para a conformidade do produto com o padrão atual.
Resumo
| Risco | Classe de Dispositivo | Auditoria QMS | Disponibilidade de Predicado | Via Regulamentar | US Agente | US Prazos da FDA |
|---|---|---|---|---|---|---|
| Baixo Risco | I | Não | N/A | Isento | Sim | 1 Mês |
| Risco Moderado | II | Sim (pós-aprovação) | Sim | PMN/510(k) | Sim | 9 - 12 Meses |
| Risco Moderado | II | Sim (pós-aprovação) | Não | Pedido de Classificação De-Novo | Sim | 18 - 30 Meses |
| Alto Risco | III | Sim (pré-aprovação) | N/A | PMA | Sim | 18 - 30 Meses |
Serviços de Registo de Dispositivos Médicos da Freyr
Experiência Freyr
- Due Diligence Regulamentar
- Documentação de Dispositivos
- Apoio 513(g)
- Registo 510(k)
- Pedido De-Novo de Classificação
- Registo PMA
- 21 CFR 820 conformidade
- Apoio a Auditorias BIMO
- MDSAP Conformidade
- Apoio à Rotulagem
- Apoio à Publicação e Submissão
- Agente US
- Reuniões de Q-Submission
- Reuniões RFD e Pré-RFD
- Certificação de Pequenas Empresas
- Registo de Estabelecimento e Listagem de Dispositivos
- Conformidade Regulatória para Dispositivos Médicos de Radiação
- Gestão de Alterações Pós-Aprovação
- Vigilância Pós-Comercialização
- Conformidade UDI
- Consultoria Regulatória para Resolução de Deficiências