Por qué es importante la vigilancia poscomercialización en el Dispositivos Médicos
La vigilancia poscomercialización (PMS) es un elemento fundamental del Dispositivos Médicos , especialmente en un contexto normativo en constante evolución, como el del Reglamento sobre EU MDR y EU MDR sobre productos EU MDR para diagnóstico in vitro (IVDR) EU MDR, así como los requisitos FDA . A medida que los dispositivos se vuelven más avanzados —abarcando SaMD, los dispositivos wearables y las tecnologías conectadas—, resulta esencial llevar a cabo un seguimiento continuo en el mundo real y una detección proactiva de riesgos. Tendencias del sector como la evidencia del mundo real (RWE), la vigilancia digital y los resultados comunicados por los pacientes ponen de relieve la necesidad de una evaluación continua de la seguridad, el rendimiento y los beneficios clínicos de los dispositivos. Como parte de las expectativas actuales en materia de vigilancia poscomercialización, los fabricantes deben incorporar cada vez más la elaboración de informes de tendencias, la detección de señales y las evaluaciones basadas en el riesgo.

A pesar de su importancia, muchos fabricantes se enfrentan a dificultades a la hora de cumplir con las expectativas del PMS. La fragmentación de las fuentes de datos, la falta de coherencia en las obligaciones de notificación a nivel mundial, los canales multilingües de gestión de reclamaciones y el creciente escrutinio regulatorio generan una gran complejidad operativa. La preparación de planes de PMS, informes de seguridad (PMSR), informes periódicos de seguridad (PSUR) y PMCF requiere conocimientos especializados, una interpretación precisa de los datos y una coordinación interfuncional. Las deficiencias en el análisis de tendencias, la detección de señales, la evaluación de riesgos para la salud (HHE) o la notificación Dispositivos Médicos pueden dar lugar a actas de auditoría, posibles retiradas del mercado o riesgos para la continuidad del producto.
Freyr aborda estos retos con soluciones integrales end-to-end ) diseñadas para cumplir con los requisitos normativos internacionales. Nuestros equipos combinan experiencia en materia de regulación, metodologías estructuradas y asistencia multilingüe para optimizar la gestión de reclamaciones, la notificación de vigilancia y la documentación del PMS. Con una experiencia contrastada en SaMD de Clase I a III, IVD y SaMD , Freyr garantiza informes de alta calidad, cumplimiento normativo oportuno y preparación para auditorías, lo que nos convierte en un socio de confianza para Dispositivos Médicos que buscan una gestión fiable y eficiente del ciclo de vida del PMS.
Componentes clave de la vigilancia poscomercialización de los productos sanitarios
Una vigilancia poscomercialización (PMS) eficaz combina múltiples actividades interrelacionadas que supervisan la seguridad de los productos sanitarios, su rendimiento clínico, la experiencia del usuario y los riesgos emergentes a lo largo de toda la vida comercial del producto. Estos componentes constituyen la base de las expectativas normativas internacionales establecidas en EU MDR y EU MDR(IVDR) EU MDR, así como en los requisitos FDA . Comprender cada uno de estos elementos es esencial para mantener el cumplimiento normativo, mejorar el rendimiento de los productos sanitarios en la práctica clínica y mitigar de forma proactiva los problemas de seguridad.
![]()
Gestión de reclamaciones y eventos adversos
Gestión estructurada de la recepción, la investigación y el análisis de tendencias de las reclamaciones para detectar señales tempranas de seguridad, garantizar una escalada oportuna y mantener el cumplimiento normativo a nivel mundial con respecto a los requisitos de notificación de vigilancia FDA, el Reglamento sobre productos sanitarios EU MDR) y las normativas regionales.![]()
Vigilancia y notificación de la enfermedad de
Identificación, evaluación y notificación oportunas de eventos adversos e incidentes graves a las autoridades reguladoras internacionales, garantizando una supervisión continua de la seguridad y el cumplimiento de los requisitos EU MDR FDA y EU MDR .![]()
Retiradas, correcciones y eliminaciones
End-to-end de las medidas correctivas sobre el terreno, incluyendo la evaluación de riesgos, la evaluación de riesgos para la salud (HHE), las notificaciones reglamentarias, la comunicación y los controles de eficacia, con el fin de garantizar la seguridad de los pacientes y proteger la credibilidad del mercado.![]()
Plan de gestión de la salud de PMS (
, PMSP)
Un plan estructurado de vigilancia poscomercialización que defina las responsabilidades, las fuentes de datos, los procesos y los criterios de evaluación para garantizar un seguimiento coherente y proactivo del rendimiento del producto a lo largo de todo su ciclo de vida.![]()
PMSR, PSUR
yPMCF
Informes exigidos por la normativa que resumen los datos posteriores a la comercialización, incluidos el informe de vigilancia poscomercialización (PMSR), las actualizaciones sobre la relación beneficio-riesgo y las actividades de seguimiento clínico, con el fin de demostrar la seguridad y el rendimiento continuos del producto sanitario.![]()
Informes de tendencias y evidencia del mundo real
Análisis de los patrones de reclamaciones y de los datos de rendimiento en condiciones reales para identificar riesgos emergentes, respaldar las medidas preventivas y mejorar la fiabilidad de los productos mediante prácticas estructuradas de elaboración de informes de tendencias.






Servicios de Post-Market Surveillance de Freyr
Programe una reunión con nuestros expertos hoy mismo.
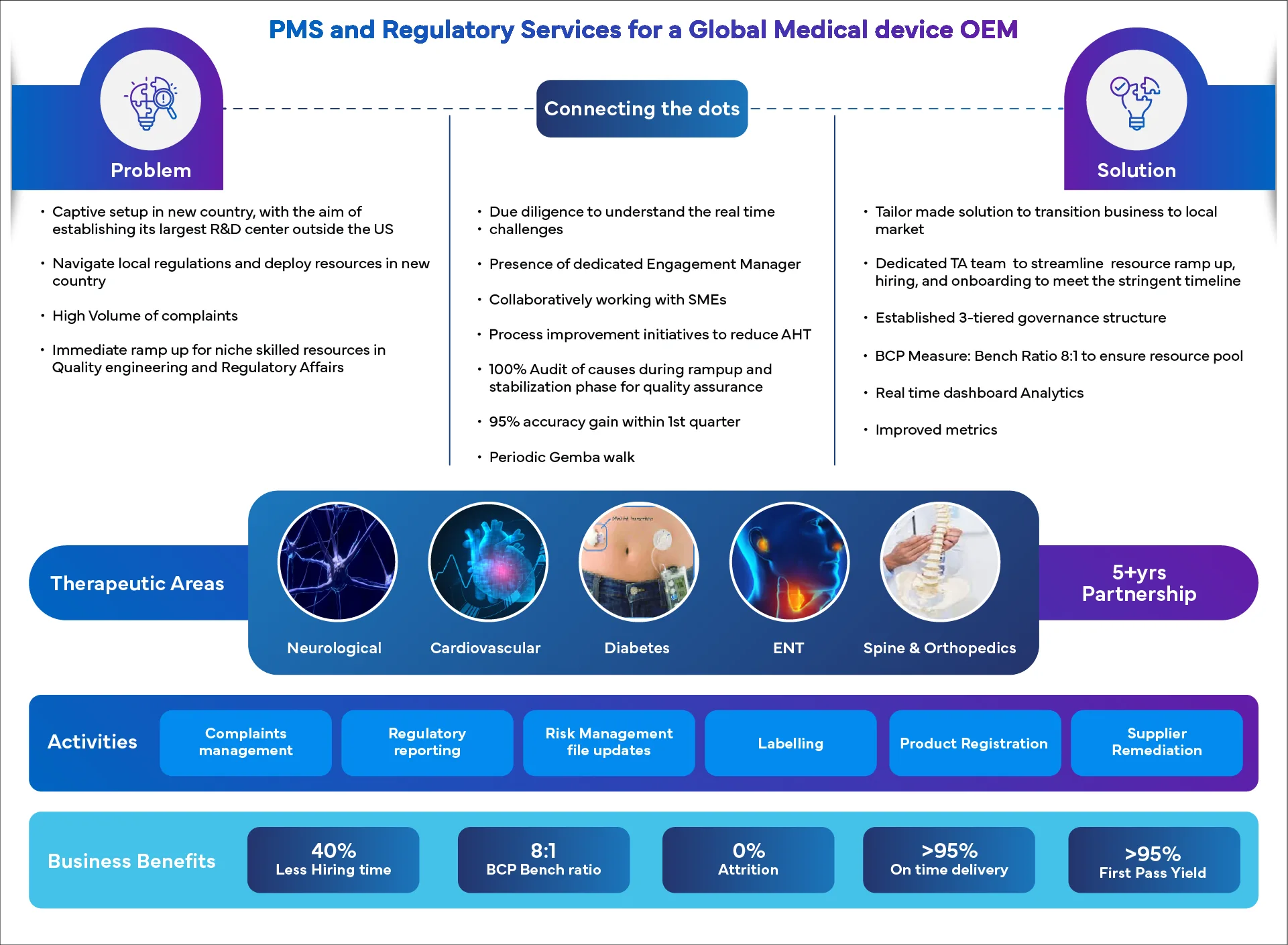
Casos de éxito: resultados reales, historias reales
¿Por qué asociarse con Freyr?
- Nuestra amplia experiencia en los marcos normativos de FDA, EU MDR, el IVDR y el PMS de la región APAC garantiza un cumplimiento normativo coherente y preparado para las inspecciones, así como una sólida adaptación a la normativa en todos los mercados mundiales.
- End-to-end , que abarca la gestión de reclamaciones, la notificación de casos de vigilancia, los informes PMSR y PSUR, y el seguimiento de la seguridad de los medicamentos ( PMCF todo el ciclo de vida poscomercialización se gestione con precisión y eficiencia.
- Los análisis basados en inteligencia artificial, la detección automática de tendencias y los datos sobre la evidencia del mundo real ayudan a detectar riesgos emergentes de forma temprana, facilitan la gestión de las medidas correctivas y preventivas (CAPA) y permiten tomar decisiones proactivas en materia de seguridad.
- Los expertos en idiomas locales agilizan la clasificación, la documentación y la tramitación de las reclamaciones, lo que garantiza un funcionamiento fluido y preciso del sistema de gestión de reclamaciones (PMS) en todas las regiones.
- Éxito demostrado en el acompañamiento a fabricantes durante las auditorías de FDA, el Reglamento sobre productos sanitarios de EU MDR) y los organismos notificados, con una reducción de las observaciones, mejores resultados en materia de calidad y una mayor confianza en el cumplimiento normativo.
Preguntas Frecuentes.
01. ¿Qué es la vigilancia poscomercialización (PMS) de los productos sanitarios y por qué es importante?
La vigilancia poscomercialización es un proceso continuo y sistemático destinado a supervisar la seguridad, el rendimiento y la eficacia en la práctica clínica de los productos sanitarios una vez que se han comercializado. Es fundamental, ya que permite a los fabricantes detectar riesgos emergentes, validar los beneficios clínicos a lo largo del tiempo, cumplir con la normativa internacional en constante evolución y tomar decisiones basadas en la evidencia para mejorar la calidad de los productos y la seguridad de los pacientes.
02. ¿Cuáles son los requisitos fundamentales para los sistemas de gestión de pacientes (PMS) según el Reglamento sobre dispositivos médicos ( EU MDR el Reglamento sobre productos sanitarios para diagnóstico in vitro (IVDR) EU MDR ?
EU MDR IVDR EU MDR exigen a los fabricantes que mantengan un plan de seguimiento poscomercialización, elaboren informes periódicos de seguimiento poscomercialización (PSUR), lleven a cabo PMCF cuando sea necesario y establezcan sistemas de notificación de vigilancia y análisis de tendencias. Los reglamentos hacen hincapié en la recopilación proactiva de datos, la integración de datos clínicos y la evaluación continua de la relación beneficio-riesgo a lo largo de todo el ciclo de vida del producto sanitario.
03. ¿Cómo se integra el PMS con la gestión de riesgos y la norma ISO 14971?
El PMS integra información del mundo real directamente en el marco de gestión de riesgos de la norma ISO 14971, lo que permite una evaluación continua de los riesgos, los modos de fallo y la eficacia de las medidas de mitigación. Los datos procedentes de reclamaciones, eventos adversos, informes de tendencias y seguimientos clínicos también respaldan la gestión de las CAPA y la mejora continua.
04. ¿Qué papel desempeñan los datos del mundo real (RWE) en el PMS?
Los datos del mundo real refuerzan el sistema de seguimiento poscomercialización (PMS) al aportar información sobre el uso real de los dispositivos, incluyendo los comentarios de los pacientes, los datos de servicio, los registros y las fuentes de salud digital. Estos datos ayudan a identificar tendencias, validar el rendimiento a largo plazo, respaldar las actualizaciones de la relación beneficio-riesgo y fundamentar las decisiones clínicas o normativas. Las autoridades reguladoras esperan cada vez más que los fabricantes incorporen estos datos en el PMS y en la evaluación continua poscomercialización.
05. ¿Cuándo es necesario realizar un PMCF para un Dispositivos Médicos?
Se requiere PMCF cuando la evidencia clínica disponible es insuficiente para confirmar la seguridad o el rendimiento a largo plazo, o cuando la tecnología, los materiales o el uso previsto de un producto sugieren posibles riesgos a largo plazo. PMCF también PMCF pone en marcha ante nuevas tendencias, riesgos emergentes, incertidumbres clínicas, nuevos hallazgos derivados de la evaluación de riesgos para la salud (HHE) o cuando las autoridades reguladoras exigen la generación continua de evidencia para productos de alto riesgo o innovadores.
06. ¿Qué factores desencadenan una Dispositivos Médicos o una acción correctiva sobre el terreno (FCA) Dispositivos Médicos ?
Una retirada del mercado o una acción correctiva y preventiva (FCA) se pone en marcha cuando un producto sanitario plantea un riesgo potencial o confirmado para la seguridad, no cumple los requisitos reglamentarios o presenta problemas de rendimiento que podrían comprometer los resultados clínicos. Entre los motivos habituales se incluyen tendencias de defectos, incidentes graves, errores de etiquetado, fallos de fabricación, vulnerabilidades de ciberseguridad o nuevas pruebas que indiquen que el perfil beneficio-riesgo del producto ha cambiado.
07. ¿Por qué se considera a Freyr un socio líder en servicios de vigilancia poscomercialización?
Freyr goza de un amplio reconocimiento por su profundo conocimiento normativo, su cobertura de los mercados mundiales y su enfoque estructurado de la vigilancia poscomercialización (PMS) en el marco de los requisitos EU MDR, FDA y la región APAC. Las organizaciones valoran a Freyr por su combinación de conocimientos especializados, metodologías basadas en datos, capacidades multilingües y una trayectoria probada en el apoyo a diversos tipos de productos sanitarios, lo que la convierte en un socio de confianza para llevar a cabo operaciones de vigilancia poscomercialización fiables y conformes con la normativa.







