
2 min di lettura
Le Autorizzazioni all'Uso di Emergenza (EUA) sono importanti per rendere i medicinali salvavita disponibili ai pazienti più rapidamente. Sono state utili per affrontare pandemie come il COVID-19. Un'iniziativa della United States Food and Drug Administration (USFDA) è iniziata nel 2004, e l'EUA è entrata in vigore quando la Sezione 564 del Federal Food, Drug, and Cosmetic Act è stata modificata dal Project BioShield Act. Questo programma è un passo intrapreso dalla FDA per proteggere la salute pubblica garantendo la sicurezza, l'efficacia e la qualità dei prodotti medici, affrontando al contempo le emergenze mediche e le minacce emergenti alla salute pubblica.
Comprendiamolo meglio nelle righe seguenti.
Comprendere l'EUA
Il percorso EUA è un mezzo per facilitare l'accessibilità delle contromisure mediche in tempi di emergenze dichiarate. Il Commissario della FDA può autorizzare quanto segue in tali situazioni:
- L'uso autorizzato di prodotti medici non approvati.
- L'uso non autorizzato di prodotti medicali approvati.
I prodotti coperti dall'EUA includono vaccini, fluidi IV, farmaci, dispositivi, test, ecc., e possono essere utilizzati per diagnosticare, trattare o prevenire condizioni potenzialmente letali. I prodotti ottengono l'EUA se sono soddisfatti i seguenti criteri:
- Prova di una condizione/malattia potenzialmente letale.
- I dati scientifici forniscono prove sufficienti che il prodotto è efficace per l'uso previsto.
- I benefici del prodotto superano i rischi (ovvero prova di sicurezza).
- Mancanza di prodotti alternativi.
Si consiglia agli sponsor di comprendere in anticipo i requisiti della FDA in modo da seguire il miglior processo normativo e garantire una presentazione EUA senza errori. Di seguito sono riportate le informazioni obbligatorie che devono essere condivise dallo sponsor nella domanda.
Dati da presentare dallo Sponsor alla FDA per un'Approvazione EUA
- Descrizione del prodotto e il suo uso previsto.
- Lo stato di approvazione del prodotto presso la FDA.
- Informazioni sulla sicurezza e l'efficacia, come dati clinici e non clinici, ecc.
- Rapporto di analisi rischio-beneficio.
- Dati di chimica, produzione e controlli (CMC).
- Informazioni sul dosaggio, controindicazioni, avvertenze ed eventi avversi per la distribuzione del prodotto medico in questione.
Come vengono rilasciate le EUA dalla FDA?
Di seguito è riportata un'interpretazione schematica del ciclo di vita dell'EUA in breve:
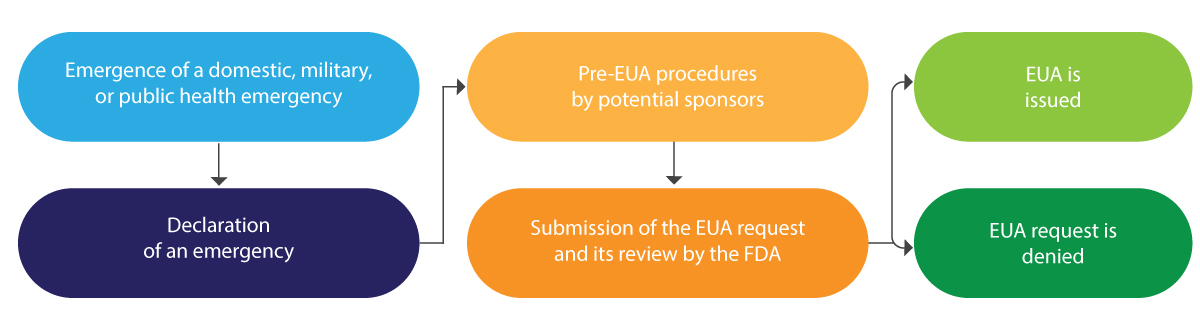
L'EUA viene tipicamente rilasciata per un periodo limitato e, al termine della suddetta emergenza, viene revocata dalla FDA.
EUA e COVID-19
Il Segretario della Salute e dei Servizi Umani (HHS) ha dichiarato il COVID-19 una pandemia il 31 gennaio 2020. Da allora, la FDA è stata fondamentale nell'approvare alcuni vaccini e kit di test domiciliari nell'ambito del percorso EUA per affrontare l'epidemia globale in corso.
Con l'emergere di nuove varianti del COVID-19, l'intera industria farmaceutica sta lavorando intensamente per frenare la diffusione e ridurre i tassi di mortalità. C'è bisogno di nuovi prodotti medicinali e di approvazioni più rapide da parte delle Autorità Regolatorie globali, in modo che il loro tempo di immissione sul mercato sia ridotto. Secondo gli esperti, il percorso EUA per la registrazione di nuovi prodotti medicinali/farmaci è la via da seguire. Diverse altre Autorità Sanitarie, come l'European Medicines Agency (EMA), la Central Drugs Standard Control Organization (CDSCO), la Saudi Food and Drug Authority (SFDA), ecc., hanno anche implementato il percorso EUA per approvazioni più rapide.
Se siete un produttore di farmaci e cercate un'EUA per il vostro prodotto salvavita, avrete bisogno di un fornitore esperto di soluzioni normative. Rivolgetevi a Freyr per un tempo di immissione sul mercato più rapido e un percorso conforme.









