2 minuty czytania
„Urządzenie referencyjne” to wyrób medyczny, który został wcześniej zatwierdzony przez US Food and Drug Administration (US FDA) i jest już na rynku, co służy jako punkt odniesienia dla nowych wyrobów medycznych ubiegających się o zatwierdzenie poprzez ścieżkę zatwierdzenia FDA 510(k).
Należy udowodnić, że dany wyrób jest co najmniej tak samo bezpieczny i skuteczny jak wyrób referencyjny pod względem jego przeznaczenia i charakterystyki technologicznej. To porównanie jest znane jako określenie „znacznej równoważności”.
Nowy wyrób nie musi być identyczny z wyrobem referencyjnym, aby był zasadniczo równoważny z wyrobem referencyjnym.
Jak zidentyfikować urządzenie referencyjne?
Baza danych FDA udostępnia trzyliterowy kod produktu dla każdej klasyfikacji wyrobu. Baza danych FDA 510(k) zawiera informacje o wszystkich wyrobach zatwierdzonych w ramach procesu 510(k). Po uzyskaniu trzyliterowego kodu produktu można uzyskać listę każdego produktu, każdej firmy oraz nazwę handlową każdego konkurenta lub potencjalnego konkurenta, którego chcesz sprawdzić. Następnie można przeprowadzić dogłębną analizę i porównanie, aby zawęzić wybór urządzenia referencyjnego.
Poniżej przedstawiono schemat blokowy przedstawiający proces identyfikacji i zawężania wyboru urządzenia referencyjnego.
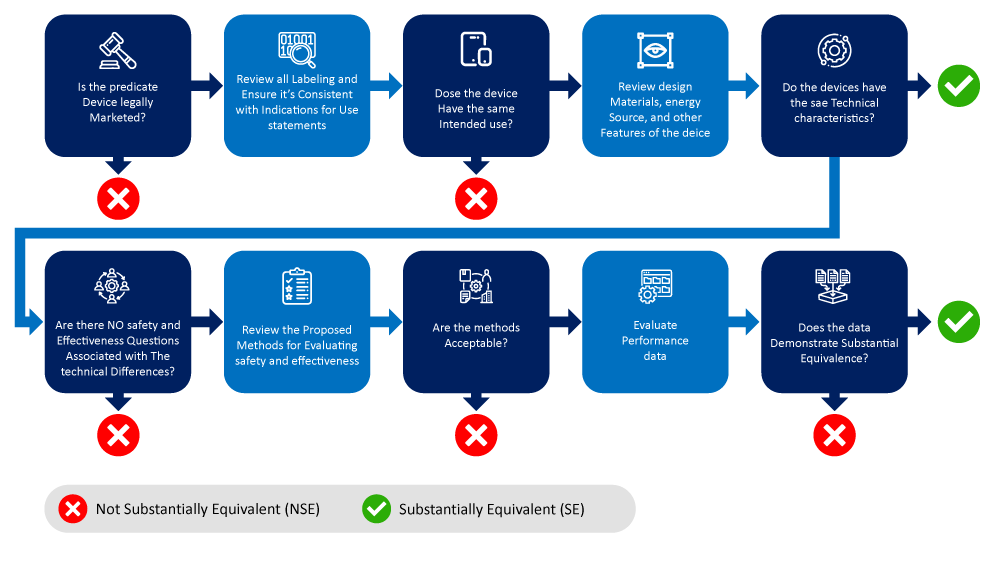
Czynniki do rozważenia przy określaniu urządzenia referencyjnego (urządzeń referencyjnych).
- Przeznaczenie: Przeznaczenie po wyrobie referencyjnym powinno być podobne do przeznaczenia nowego wyrobu. Na przykład, jeśli nowy wyrób jest przeznaczony do monitorowania pracy serca, wyrób referencyjny również powinien być wyrobem do monitorowania pracy serca.
- Charakterystyka technologiczna: Urządzenie referencyjne powinno być identyczne z nowym urządzeniem pod względem cech technologicznych. Na przykład, projekt, użyte materiały i metoda działania powinny być podobne.
- Biokompatybilność: Oceny biokompatybilności wyrobu medycznego lub jego komponentu nie powinny ograniczać się do surowców użytych w wyrobie i procesie produkcyjnym; należy również uwzględnić dodatkowe substancje chemiczne. Czynnik ten nie dotyczy jednak wyrobów medycznych do diagnostyki in vitro (IVD).
- Najnowsza technologia: Urządzenie referencyjne nie powinno być przestarzałe i powinno reprezentować najnowszą technologię medyczną.
Urządzenie referencyjne jest kluczowym czynnikiem w określaniu, czy nowy wyrób medyczny może zostać wprowadzony na rynek poprzez ścieżkę 510(k). Wybór niewłaściwego urządzenia referencyjnego może skutkować droższym i bardziej czasochłonnym procesem zatwierdzania regulacyjnego, natomiast wybór odpowiedniego urządzenia referencyjnego może pomóc zmniejszyć koszty i czas potrzebny na wprowadzenie nowego wyrobu medycznego na rynek. Jeśli urządzenie referencyjne nie jest odpowiednie, może to prowadzić do opóźnień i dodatkowych kosztów.
Aby uzyskać pomoc w procesie składania wniosku 510(k) dla wyrobu medycznego, umów się na rozmowę z ekspertami regulacyjnymi Freyr, którzy pomogą Ci przejść przez procedury. Bądź na bieżąco. Zachowaj zgodność.









