
3 min de lectura
En el sector Dispositivos Médicos , en constante evolución y altamente regulado, garantizar la calidad de los productos y la seguridad de los pacientes no es solo una obligación legal, sino también un imperativo estratégico. Unos sistemas eficaces de gestión de reclamaciones y de vigilancia, de conformidad con ISO 13485:2016, constituyen la columna vertebral de cualquier sistema de gestión de la calidad (SGC) sólido. Estos mecanismos garantizan el cumplimiento de la normativa internacional y refuerzan la confianza de los clientes y la credibilidad de la marca.
Analicemos cómo la gestión de reclamaciones y la vigilancia desempeñan un papel fundamental a la hora de garantizar la calidad y el cumplimiento normativo en el Dispositivos Médicos , con información basada en ISO 13485:2016 y respaldada por los marcos normativos internacionales.
Comprensión de la gestión de reclamaciones según la norma ISO 13485:2016
ISO 13485:2016 define una reclamación como cualquier comunicación escrita, oral o electrónica en la que se aleguen defectos relacionados con la identidad, la calidad, la durabilidad, la fiabilidad, la facilidad de uso, la seguridad o el rendimiento Dispositivos Médicos. Esta norma exige a Dispositivos Médicos que establezcan procedimientos documentados para la gestión de reclamaciones, lo que incluye la evaluación, la investigación y la resolución de las mismas, como parte de un sistema eficaz de gestión de la calidad.
Un sistema de gestión de reclamaciones es fundamental para garantizar el cumplimiento normativo, mantener la satisfacción del cliente y mejorar la calidad de los dispositivos. El proceso debe estar bien organizado y ser transparente, e incluir los siguientes pasos:
- Recepción y documentación de reclamaciones
Todas las reclamaciones deben registrarse sin demora, incluyendo el mayor número posible de detalles sobre la naturaleza del problema, las especificaciones y el modelo del dispositivo, las condiciones de uso y cualquier consecuencia que haya sufrido el usuario o el paciente. - Evaluación de la obligación de notificación
Una vez registradas, las reclamaciones deben evaluarse para determinar si se trata de incidentes que deben notificarse. Por ejemplo, en EE. UU., Dispositivos Médicos están obligados a notificar determinados problemas relacionados con los dispositivos en el marco del sistema Dispositivos Médicos (MDR) regulado por la FDA. Los fabricantes deberán cumplir la normativa aplicable en materia de plazos de notificación. - Investigación y análisis de las causas fundamentales
Las reclamaciones fundadas deben ir seguidas de una investigación detallada. Identificar la causa fundamental es esencial para aplicar medidas correctivas y preventivas (CAPA) eficaces. - Medidas correctivas y preventivas (CAPA)
: A la luz de Dispositivos Médicos , las empresas deben poner en marcha medidas CAPA para resolver tanto el problema inmediato como las recurrencias y evitar que se repitan en el futuro. - Bucle de retroalimentación y cierre
Una vez resuelto el problema, la reclamación debe cerrarse formalmente con la documentación completa y se debe informar al reclamante, si procede en el caso de los Dispositivos Médicos.
Vigilancia: un pilar fundamental de la supervisión posterior a la comercialización
La vigilancia consiste en el seguimiento y la notificación de eventos e incidentes adversos una vez que un Dispositivos Médicos comercializado. De conformidad con ISO 13485:2016 y la normativa internacional correspondiente, esta vigilancia continua es esencial para la detección precoz de posibles riesgos y la aplicación de medidas correctivas oportunas.
El sistema de vigilancia de la Unión Europea, tal y como se ha actualizado en virtud del Dispositivos Médicos (EU MDR), exige Dispositivos Médicos que dispongan de planes de vigilancia poscomercialización. Estos planes deben incluir procesos para recopilar y examinar datos sobre el uso real de los productos sanitarios.
Del mismo modo, en la India, la Central Drugs Standard Control Organization CDSCO) regula los procesos de vigilancia y tramitación de reclamaciones relativos a los productos sanitarios. Fomenta la notificación de acontecimientos adversos y fallos de los productos con el fin de mejorar continuamente Dispositivos Médicos y la eficacia Dispositivos Médicos .
Entre los elementos clave de un sistema de vigilancia eficaz se incluyen:
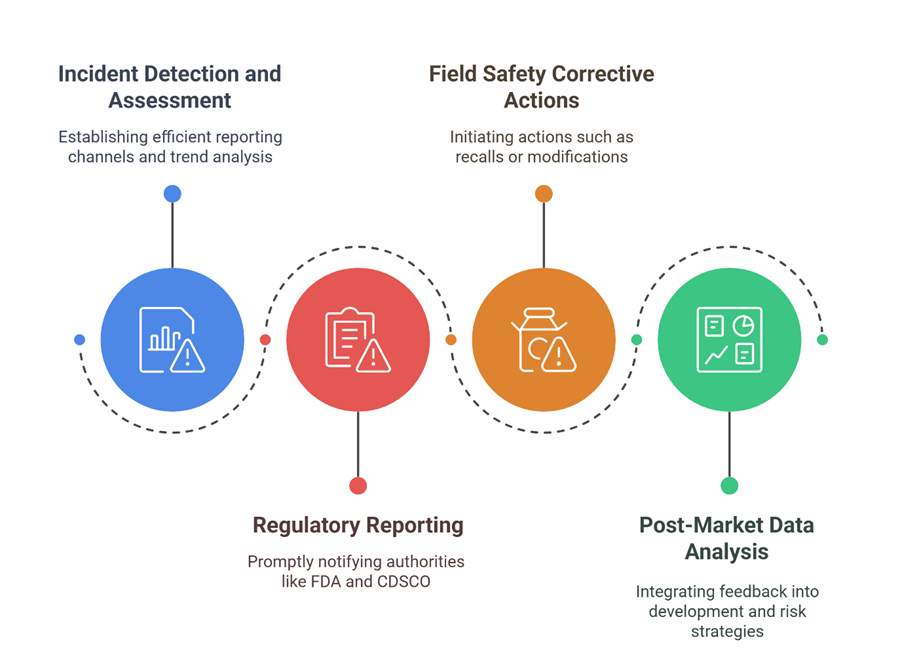
Elaboración de una estrategia integrada de gestión de reclamaciones y vigilancia
Dispositivos Médicos pueden mejorar su sistema de gestión de la calidad integrando la gestión de reclamaciones y la vigilancia en un único sistema coherente. A continuación se presentan las mejores prácticas:
Procedimientos operativos estándar (SOP) y definiciones de funciones claras
Los procedimientos operativos estándar (SOP) deben describir los pasos para la recepción, evaluación, notificación y resolución de reclamaciones. En ellos se establece quién es el responsable en cada fase para evitar retrasos y lagunas.
- Formación y sensibilización del personal
La formación periódica del personal garantiza que los empleados sepan identificar y gestionar de forma eficaz las quejas y los incidentes adversos. - Herramientas digitales y automatización
El uso del software QMS permite agilizar el seguimiento de las reclamaciones, la documentación y la elaboración de informes. La automatización mejora la trazabilidad y el cumplimiento de ISO 13485:2016. - Cultura de la calidad y mejora continua
Promover una filosofía que valore la retroalimentación fomenta la notificación proactiva de problemas y estimula la innovación en la mejora de la calidad.
El cumplimiento normativo y más allá: una ventaja competitiva en marketing
La implantación de sistemas conformes con ISO 13485:2016 para la gestión de reclamaciones y la vigilancia no solo garantiza el cumplimiento normativo, sino que también posiciona a la empresa como una entidad fiable y centrada en la calidad. En un sector en el que la seguridad y el rendimiento repercuten directamente en la vida de las personas, ese compromiso puede constituir una poderosa herramienta de marketing.
Según FDA el cumplimiento de las normas internacionales mejora el acceso a los mercados mundiales, refuerza la confianza de los clientes y reduce el riesgo de costosas retiradas de productos o litigios.
| Región | Marco Reglamentario | N.º de referencia/artículo para la tramitación de reclamaciones | N.º de cláusula de referencia / Artículo sobre vigilancia / Notificación de acontecimientos adversos |
| Unión Europea | EU MDR 2017/745 | Artículo 83, anexo III (sistema PMS); artículo 10, apartado 9 | Artículos 87 a 92 (Sistema de vigilancia), anexo III |
| Estados Unidos (FDA) | 21 CFR, parte 820 (QSR) | §820.198 – Expedientes de reclamaciones | 21 CFR, parte 803: Dispositivos Médicos (MDR) |
| India (CDSCO) | Dispositivos Médicos de 2017 (MDR) | Capítulo VII, Reglas 25 y 26 | Capítulo VII, Regla 27 |
| Internacional (norma ISO) | ISO 13485:2016 | Cláusula 8.2.2 – Gestión de reclamaciones | Cláusula 8.2.3 – Notificación a las autoridades reguladoras |
Conclusión
En el competitivo panorama Dispositivos Médicos actual Dispositivos Médicos , es imprescindible contar con sistemas eficaces de gestión de reclamaciones y vigilancia. En consonancia con ISO 13485:2016, estos procesos no solo protegen a los pacientes, sino que también contribuyen a construir una marca sólida y centrada en la reputación. Al invertir en procedimientos sistemáticos, formación continua y vigilancia poscomercialización, los fabricantes pueden convertir los requisitos normativos en oportunidades de crecimiento y liderazgo en el mercado.









