
3 minuty czytania
Protokół walidacji jest zdefiniowany jako udokumentowany plan testowania wyrobu medycznego w celu potwierdzenia, że proces produkcyjny użyty do wytworzenia produktu spełnia specyficzne wymagania użytkownika, techniczne i regulacyjne. Obejmuje to przegląd zmiennych procesowych i ograniczeń operacyjnych oraz analizę wyników testów w rzeczywistych warunkach użytkowania.
Proces walidacji obejmuje kilka konkretnych działań. Kroki te przedstawiono w następujący sposób:
- Najpierw tworzy się zespół walidacyjny, a każdemu członkowi przypisuje się określone role i obowiązki. Celem walidacji procesu jest przedstawienie jasnego oświadczenia o celach walidacji i zdefiniowanie zakresu działań walidacyjnych poprzez określenie aspektów wyrobu medycznego, które są walidowane. Następnie zespół rozumie podstawowe zasady procesu, aby zidentyfikować konkretne parametry i pożądane wyniki.
- Po drugie, ustala się kryteria oceny i akceptacji, a także wybiera odpowiednie metody testowania, narzędzia i techniki analizy statystycznej. Następnie opracowuje się protokoły walidacji procesu oraz wdraża kwalifikację instalacyjną (IQ), kwalifikację operacyjną (OQ) i kwalifikację działania (PQ).
- Na koniec, określa się bieżące kontrole procesów i środki monitorowania, aby zapewnić ciągłą walidację procesu. W razie potrzeby przeprowadzana jest rewalidacja w celu utrzymania dokładności i skuteczności procesu walidacji.
Rysunek 1 poniżej przedstawia krok po kroku proces walidacji.
Rysunek 1: Etapy procesu walidacji
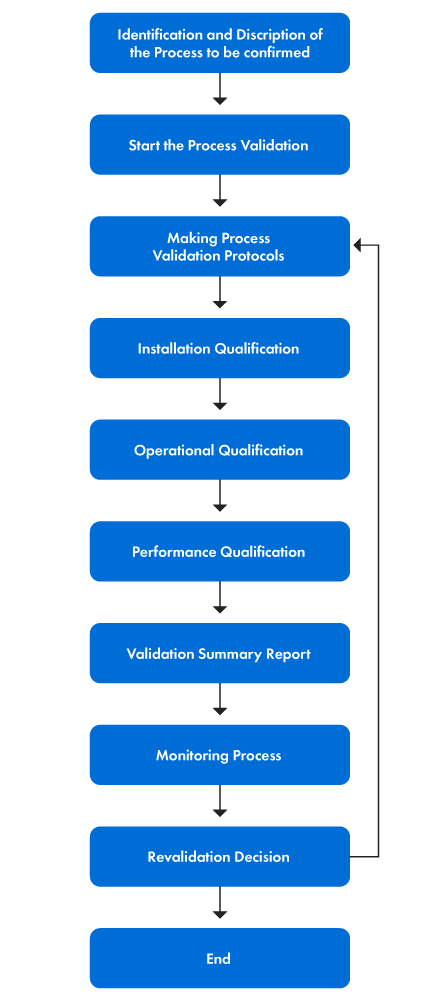
PVP
Ze względu na szeroki zakres wolumenów produkcji i złożoności wytwarzania, istnieje wiele podejść do przeprowadzania walidacji procesu. Jednak przepisy Amerykańskiej Agencji ds. Żywności i Leków (USFDA) oraz ISO 13485 zawierają ograniczone sugestie dotyczące konkretnych metod. Niemniej jednak, szeroko uznanym i autorytatywnym źródłem dla walidacji procesów wyrobów medycznych jest dokument wytycznych Global Harmonization Task Force (GHTF), obecnie nazywanego International Medical Device Regulators Forum (IMDRF), opublikowany w 2004 roku. Pozostaje on głównym odniesieniem nawet na oficjalnej stronie internetowej USFDA.
Zgodnie z dokumentem wytycznych, powołuje się zespół walidacyjny w celu stworzenia szczegółowego Planu Walidacji Procesu (PVP). Protokoły walidacji procesu zawierają szczegółowy schemat wdrażania IQ, OQ, PQ i rewalidacji. PVP powinien zawierać następujące elementy:
- Definiowanie wyrobu i określanie podejścia do walidacji.
- Identyfikacja elementów wymagających walidacji.
- Prowadzenie działań w wyznaczonym miejscu.
- Określenie zakresu dokumentacji.
- Tworzenie harmonogramu dla działań walidacyjnych.
- Opracowywanie ogólnego harmonogramu głównego.
- Utrzymywanie kompleksowej listy i odniesień do przeprowadzonych walidacji wewnętrznych i zewnętrznych.
Protokół walidacji jest sporządzany przed rozpoczęciem działań walidacyjnych. Powinien być przygotowany przez zespół walidacyjny i zatwierdzony przez odpowiedni dział. Celem protokołu walidacji jest określenie scenariuszy testowych, które należy wykonać, aby zagwarantować, że procesy i sprzęt są gotowe do wytwarzania bezpiecznych i skutecznych wyrobów medycznych.
Raport analityczny, który zawiera informacje wraz z niezbędnymi analizami, wyjaśnieniami i zaleceniami, stanowi część protokołu walidacji. Zapisy te są następnie weryfikowane, aby upewnić się, że spełnione są następujące dwa (02) kryteria:
- Spełnienie norm regulacyjnych.
- Wszystkie wygenerowane zapisy i dane są przeglądane pod kątem wyników, adekwatności i kompletności.
Poniższy rysunek 2 przedstawia PVP oraz różne procesy z nim związane.
Rysunek 2: PVP i jego wymagania
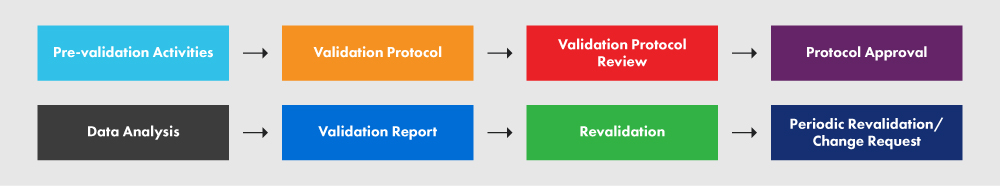
Odpowiednio sporządzony protokół zawiera jasne wytyczne, zasady i procedury, których należy przestrzegać podczas walidacji procesu. Obejmuje on takie aspekty jak obiekty, sprzęt, metody i szkolenia. Protokół określa dane wejściowe i granice procesu, a także kluczowe kroki do pomyślnego wykonania projektu walidacji procesu. Chociaż poniższy zarys nie obejmuje każdego pojedynczego elementu wymaganego w protokole, daje on ogólny obraz wymaganego poziomu szczegółowości. Zdecydowanie zalecamy zapoznanie się z dokumentem wytycznych w celu lepszego zrozumienia procesu.
- Strona tytułowa
- Produkty objęte zakresem
- Sprzęt/proces do walidacji
- Ogólne
- Cele
- Dokumenty referencyjne
- Plan Walidacji
- IQ
- OQ
- PQ
- Sprzęt pomiarowo-testowy i kalibracja
- Konserwacja sprzętu
- Revalidacja
- Strona zatwierdzenia/podpisu zespołu walidacyjnego
Zarządzanie operacjami odgrywa kluczową rolę w utrzymaniu optymalnej wydajności poprzez monitorowanie kluczowych wskaźników, przegląd metod i procedur pracy oraz podejmowanie szybkich działań w przypadku pojawienia się problemów. W sytuacjach problematycznych może być konieczne częściowe lub nawet pełne ponowne zatwierdzenie procesu. Zgodnie z Sekcją 820.75(c) Rozporządzenia USFDA dotyczącego Systemu Jakości (QSR), ponowna walidacja procesu powinna być rozważona w następujących okolicznościach: „W przypadku wystąpienia zmian lub odchyleń w procesie, producent musi dokonać przeglądu, oceny i, w stosownych przypadkach, przeprowadzić ponowną walidację. Działania te muszą być udokumentowane.”
Możliwe czynniki wywołujące rewalidację procesu obejmują modyfikacje specyfikacji, metod, procedur, oprogramowania, projektów, kluczowych komponentów, skalowania partii, zmiany lokalizacji, zmiany sprzętu i tym podobne. Ponadto, wdrożenie działań korygujących i zapobiegawczych (CAPA) może również służyć jako czynnik wywołujący rewalidację procesu. Główne powody rewalidacji są następujące:
- Zmiany wprowadzone w procesie.
- Negatywny trend w jakości, nagłe pogorszenie jakości lub gwałtowny wzrost skarg klientów.
- Znaczące rozszerzenie zdolności produkcyjnych linii.
- Zmiany w projekcie.
- Zmiany w opakowaniu produktu.
- Przeniesienie procesu do innej placówki.
- Zmiany w procesie składania wniosków.
Aby dowiedzieć się więcej o protokołach walidacji i ich znaczeniu w produkcji wyrobów medycznych, skonsultuj się z nami. Bądź na bieżąco! Zachowaj zgodność!









