

3 min de lectura
En el mundo en evolución de los Dispositivos Médicos, el cumplimiento no es una tarea única, es un compromiso continuo. El monitoreo continuo y las actualizaciones de informes clave como los Informes de Evaluación Clínica (CER), los Informes de Evaluación de Desempeño (PER) y los Informes Periódicos de Actualización de Seguridad (PSUR) son críticos a lo largo de todo el ciclo de vida de un Dispositivo Médico, desde la investigación inicial hasta la vigilancia post-comercialización. A medida que el panorama de los avances médicos y los requisitos reglamentarios evolucionan continuamente, garantizar la seguridad y el cumplimiento de los Dispositivos Médicos e IVD a través de una redacción médica eficaz sigue siendo la piedra angular del éxito y la viabilidad a largo plazo.
Veamos cómo la gestión del ciclo de vida sigue siendo esencial para el éxito de un Dispositivo Médico.
El lanzamiento de un dispositivo médico es la culminación de años de esfuerzo dedicados a varias fases como la investigación, el desarrollo, los ensayos clínicos, las presentaciones reglamentarias y la vigilancia post-comercialización. Este proceso abarca muchos años y cada fase acumula una gran cantidad de datos vitales que deben ser cuidadosamente recopilados y analizados para garantizar que el dispositivo siga siendo seguro.
El cumplimiento no es algo puntual, y una gestión eficaz del ciclo de vida ayuda a evitar retrasos que pueden resultar costosos más adelante. Estos retrasos suelen producirse debido a una planificación ineficaz para cambios reglamentarios imprevistos o a un incumplimiento que puede haberse pasado por alto. Las actualizaciones periódicas de documentos críticos como los CER, PER y PSUR garantizan que los fabricantes cumplan con los requisitos reglamentarios en evolución y mantengan la seguridad del producto. La gestión del ciclo de vida garantiza que los fabricantes, desde el principio, hayan planificado cada etapa, y así puedan evitar cualquier obstáculo y asegurar una entrada exitosa en el mercado y la longevidad de sus dispositivos.
¿Se mantiene al día con el cumplimiento?
Desde vendajes hasta implantes, la industria de Dispositivos Médicos es un sector vanguardista y creativo con una gran cantidad de oportunidades. A pesar del fuerte deseo de los fabricantes de ofrecer al mercado productos seguros y de alta calidad, la ambigüedad persiste. El EU MDR reemplazó la Directiva de Dispositivos Médicos (MDD) y la Directiva de Dispositivos Médicos Implantables Activos (AIMDD). Esta regulación se implementó para imponer controles más rigurosos, mejorar la seguridad del paciente y promover una mayor transparencia dentro del sector de la salud. Existe mucha incertidumbre sobre esos estándares, y con frecuencia se pasan por alto cuestiones cruciales de cumplimiento. Sin embargo, los fabricantes de Dispositivos Médicos deben tomar medidas para evitar dificultades reglamentarias.
Investiguemos los desafíos más comunes a los que se enfrentan los fabricantes de Dispositivos Médicos.
- Mantenimiento de CAPA (Corrective and Preventive Action)
- Cumplir con los procedimientos de reclamaciones
- Seguimiento de los Procedimientos de Vigilancia
- Evaluación Clínica y Vigilancia Post-Comercialización
- Conexión con organismos notificados
Mejores prácticas para mantener el cumplimiento
Para superar estos desafíos, en Freyr hemos establecido
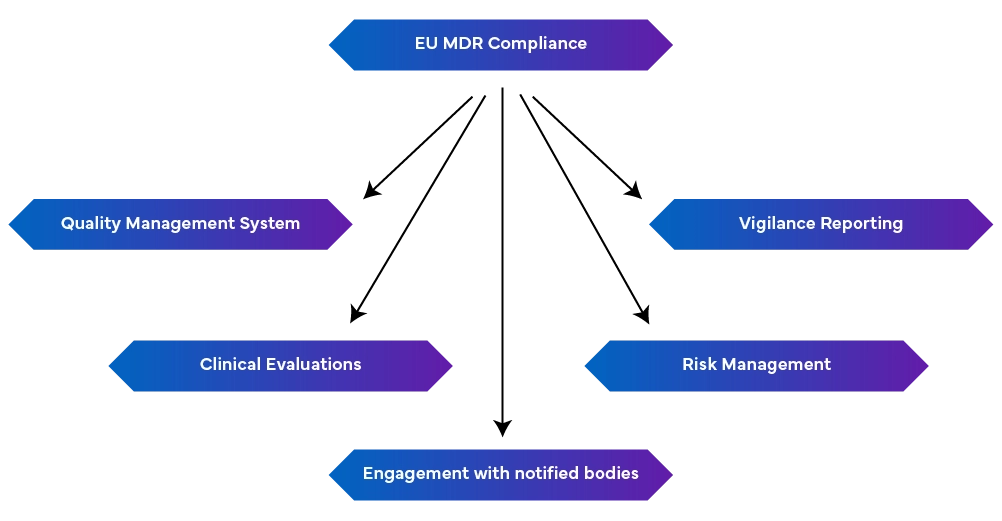
-un Sistema de Gestión de Calidad Robusto (EN ISO 13485:2016): Esto incluye aspectos de producción, como el cumplimiento reglamentario, la documentación técnica, las declaraciones de conformidad de la UE y la gestión de riesgos.
-Notificación de Vigilancia: Mantener la notificación de vigilancia como un proceso continuo en lugar de un esfuerzo puntual.
- Gestión Proactiva de Riesgos: Esto identifica y mitiga los riesgos a lo largo del ciclo de vida del dispositivo mediante actualizaciones periódicas para abordar los riesgos emergentes.
- Realizar evaluaciones clínicas y vigilancia post-comercialización: Esto se logra demostrando la seguridad y el rendimiento del dispositivo utilizando datos clínicos. Si es necesario, se deben realizar investigaciones clínicas, y las evaluaciones clínicas deben actualizarse regularmente con datos de vigilancia post-comercialización. También se requieren informes periódicos de actualización de seguridad que resuman los hallazgos.
- Relación con Organismos Notificados: Los cambios en la normativa pueden afectar los requisitos del MDR, lo que hace crucial mantener una buena relación con los Organismos Notificados para recibir actualizaciones y modificaciones a tiempo.
Cómo puede ayudarle un experto reglamentario
Mantenerse al día con un panorama reglamentario en constante cambio puede ser difícil. ¿Por qué no dejar que un experto le guíe a través de este laberinto? Gestionar las exigencias reglamentarias del ciclo de vida de un Dispositivo Médico no solo es complejo, sino que también requiere muchos recursos. En el caso de empresas más pequeñas, donde los recursos internos pueden ser mejor utilizados en otras áreas, la ayuda de un socio reglamentario externo puede resultar indispensable. Ofrecen conocimientos especializados sobre las regulaciones en evolución y aseguran que sus informes clave —como CERs, PERs y PSURs— se actualicen continuamente y cumplan con la normativa.
Puede evitar retrasos costosos, prevenir el incumplimiento y agilizar su proceso de aprobación. Además, un experto reglamentario puede ofrecer soluciones personalizadas basadas en las necesidades específicas de su dispositivo, asegurando que la vigilancia post-comercialización, el análisis de brechas y toda la documentación de cumplimiento estén actualizados. Su participación puede simplificar el proceso general, minimizar los riesgos y asegurar que el dispositivo pueda cumplir las expectativas de seguridad y rendimiento.
Freyr puede ayudarle con todas sus necesidades reglamentarias relacionadas con la gestión del ciclo de vida. ¡Póngase en contacto con nosotros hoy mismo!









