

3 min di lettura
Nel mondo in evoluzione dei dispositivi medici, la conformità non è un compito una tantum, ma un impegno costante. Il monitoraggio continuo e gli aggiornamenti di rapporti chiave come i Rapporti di Valutazione Clinica (CER), i Rapporti di Valutazione delle Prestazioni (PER) e i Rapporti Periodici di Aggiornamento sulla Sicurezza (PSUR) sono fondamentali per l'intero ciclo di vita di un dispositivo medico, dalla ricerca iniziale alla sorveglianza post-commercializzazione. Poiché il panorama dei progressi medici e dei requisiti normativi si evolve continuamente, garantire la sicurezza e la conformità dei dispositivi medici e dei IVD attraverso una scrittura medica efficace continua a essere la pietra angolare del successo e della redditività a lungo termine.
Esaminiamo come la gestione del ciclo di vita rimanga essenziale per il successo di un dispositivo medico
Il lancio di un dispositivo medico è il culmine di anni di sforzi dedicati a diverse fasi come ricerca, sviluppo, studi clinici, presentazioni normative e sorveglianza post-commercializzazione. Questo processo si estende per molti anni e ogni fase accumula dati vitali in grande quantità, che devono essere attentamente compilati e analizzati per garantire che il dispositivo rimanga sicuro.
La conformità non è un evento una tantum e una gestione efficace del ciclo di vita aiuta a evitare ritardi che potrebbero essere costosi in seguito. Questi ritardi si verificano spesso a causa di una pianificazione inefficace per cambiamenti normativi imprevisti o non conformità, che potrebbero essere stati trascurati. Aggiornamenti regolari di documenti critici come CER, PER e PSUR garantiscono che i produttori soddisfino i requisiti normativi in evoluzione e mantengano la sicurezza del prodotto. La gestione del ciclo di vita assicura che i produttori, fin dall'inizio, abbiano pianificato ogni fase, e quindi possano evitare eventuali insidie e garantire un ingresso di successo sul mercato e la longevità dei loro dispositivi.
Siete al passo con la conformità?
Dalle bende agli impianti, l'industria dei dispositivi medici è un settore all'avanguardia e creativo con una ricchezza di prospettive. Nonostante il forte desiderio dei produttori di fornire al mercato prodotti sicuri e di alta qualità, l'ambiguità persiste. L'EU MDR ha sostituito la Direttiva sui Dispositivi Medici (MDD) e la Direttiva sui Dispositivi Medici Impiantabili Attivi (AIMDD). Questa regolamentazione è stata implementata per imporre controlli più rigorosi, migliorare la sicurezza dei pazienti e promuovere una maggiore trasparenza nel settore sanitario. C'è molta incertezza riguardo a tali standard e le questioni cruciali di conformità sono spesso trascurate. Tuttavia, i produttori di dispositivi medici dovrebbero adottare misure per evitare difficoltà normative.
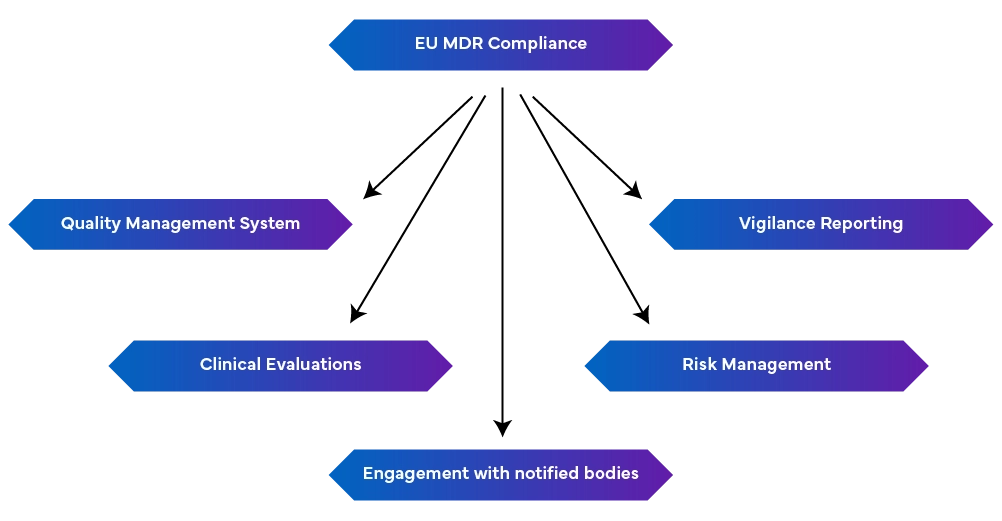
Analizziamo le sfide più comuni affrontate dai produttori di dispositivi medici.
- Mantenere la CAPA (Corrective and Preventive Action)
- Rispetto delle procedure di gestione dei reclami
- In conformità con le procedure di vigilanza
- Valutazione clinica e Sorveglianza post-commercializzazione
- Connettersi con gli organismi notificati
Migliori pratiche per rimanere conformi
Per superare queste sfide, noi di Freyr abbiamo istituito
-un solido sistema di gestione della qualità (EN ISO 13485:2016): Questo include aspetti di produzione, tra cui la conformità normativa, la documentazione tecnica, le dichiarazioni di conformità UE e la gestione del rischio.
-Segnalazione di Vigilanza: Mantenere la segnalazione di vigilanza come un processo continuo piuttosto che uno sforzo isolato.
- Gestione Proattiva del Rischio: Questo identifica e mitiga i rischi durante l'intero ciclo di vita del dispositivo, aggiornando regolarmente per affrontare i rischi emergenti.
- Condurre valutazioni cliniche e sorveglianza post-commercializzazione: Ciò si ottiene dimostrando la sicurezza e le prestazioni del dispositivo utilizzando dati clinici. Se necessario, devono essere condotte indagini cliniche e le valutazioni cliniche devono essere regolarmente aggiornate con i dati di sorveglianza post-commercializzazione. Sono inoltre richiesti rapporti periodici di aggiornamento sulla sicurezza che riassumono i risultati.
- Connessione con gli Organismi Notificati: I cambiamenti nelle normative possono influenzare i requisiti MDR, rendendo cruciale mantenere un buon rapporto con gli Organismi Notificati per aggiornamenti e modifiche tempestivi.
Come un esperto normativo può aiutarti
Mantenere il passo con un panorama normativo in continua evoluzione può essere difficile. Perché non lasciare che un esperto vi guidi attraverso questo labirinto? Gestire le esigenze normative del ciclo di vita di un dispositivo medico non è solo complesso, ma richiede anche molte risorse. Nel caso di aziende più piccole, dove le risorse interne potrebbero essere meglio utilizzate in altre aree, l'aiuto di un partner normativo esterno può rivelarsi indispensabile. Essi offrono conoscenze specializzate sulle normative in evoluzione e assicurano che i vostri rapporti chiave, come CER, PER e PSUR, siano continuamente aggiornati e conformi.
È possibile evitare ritardi costosi, prevenire la non conformità e ottimizzare il processo di approvazione. Inoltre, un esperto normativo può offrire soluzioni personalizzate in base alle esigenze specifiche del vostro dispositivo, garantendo che la sorveglianza post-commercializzazione, l'analisi delle lacune e tutta la documentazione di conformità siano aggiornate. Il loro coinvolgimento può semplificare il processo complessivo, minimizzare i rischi e garantire che il dispositivo possa soddisfare le aspettative di sicurezza e prestazioni.
Freyr può aiutarvi con tutte le vostre esigenze normative relative alla gestione del ciclo di vita. Contattateci oggi stesso!









