3 min de lectura
El etiquetado es una parte integral de la comercialización de Dispositivos Médicos. La etiqueta es una información que se adjunta al dispositivo y/o al embalaje en un formato legible para el ser humano. El propósito principal del etiquetado es proporcionar información de seguridad a los usuarios, que pueden ser profesionales de la salud, consumidores o cualquier otra persona relevante.
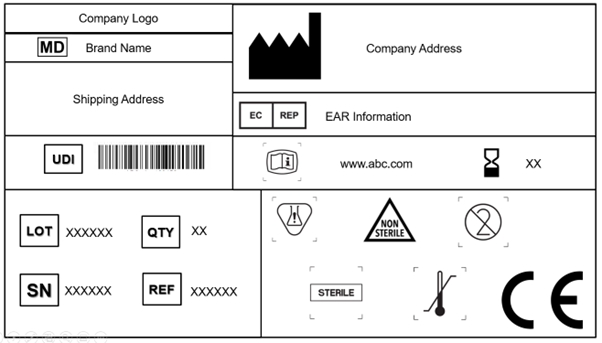
Todas las autoridades reglamentarias globales tienen ciertos requisitos de etiquetado. Asimismo, la UE ha detallado los requisitos de etiquetado en el Capítulo III del Anexo I del Reglamento de Dispositivos Médicos de la UE (EU MDR) 2017/745. Lo más importante a tener en cuenta es incluir todos los símbolos que cubren la información requerida en el etiquetado del dispositivo y en los documentos (folletos, manuales, IFU, etc.) que lo acompañan.
Algunas de las consideraciones críticas de etiquetado a tener en cuenta al cumplir con el EU MDR 2017/745 son -
1. Simbología de etiquetado de Dispositivos Médicos
Se exige a cada fabricante que incorpore el símbolo de Dispositivo Médico, que indica que el producto suministrado al mercado de la UE es un Dispositivo Médico. Es obligatorio fijar este símbolo en el dispositivo y en todos los niveles de embalaje. Además, la etiqueta debe mostrar el nombre comercial y el nombre original del dispositivo.
2. Dispositivos especiales
En caso de que el producto sea un dispositivo especial o personalizado, su estado debe mencionarse en el etiquetado. Por ejemplo, si la intención del producto es únicamente para investigación clínica, la etiqueta debe indicarlo explícitamente.
Para los dispositivos con materiales absorbentes o que puedan dispersarse localmente en el cuerpo humano, el etiquetado debe mencionar la composición del material y detalles cuantitativos sobre los componentes clave.
Incluso se requiere un etiquetado explícito en el caso de dispositivos de un solo uso y dispositivos estériles. Para los dispositivos reprocesados, el etiquetado debe mencionar el número de veces que pueden ser reprocesados, el número de veces que han sido reprocesados hasta el momento y el método de esterilización utilizado.
3. Presencia de sustancias tóxicas
La declaración de la presencia de sustancias CMR (carcinogénicas, mutagénicas, tóxicas para la reproducción) y sustancias que alteran el sistema endocrino es obligatoria en las etiquetas si la concentración es superior al 0,1% p/p. La lista de dichas sustancias debe adjuntarse al dispositivo y/o al embalaje.
Además, debe fijarse a los dispositivos una etiqueta sobre la presencia de derivados de sangre y tejidos (incluso cuando estén contenidos en la sustancia medicinal del dispositivo combinado).
4. Normas armonizadas
El EU MDR 2017/745 reconoce y acepta la norma ISO 15223-1: 2021. El documento determina los símbolos que se utilizarán en el etiquetado de los Dispositivos Médicos y su embalaje. El Capítulo 3 (23.1,h) del Anexo I del EU MDR especifica que se pueden utilizar símbolos reconocidos internacionalmente, y en el caso de regiones donde estos símbolos no sean reconocidos, se requiere que la descripción de los mismos se proporcione en un documento junto con el dispositivo.
5. UDI
Los artículos 27, 28, 29 y el Anexo VI (A, B, C) detallan las normas y reglamentos para el UDI. Ahora se requiere que la etiqueta contenga un portador UDI [Identificación Automática para la Captura de Datos (AIDC) y representación de Interpretación Legible por Humanos (HRI) del UDI] en el dispositivo y también en los niveles de embalaje superiores. El embalaje superior del dispositivo (excluyendo los paquetes de envío) tendrá su propio portador UDI.
6. Información electrónica para el uso (eIFU)
La dirección web (URL) en forma de eIFUs también puede colocarse en el etiquetado del Dispositivo Médico junto con las IFUs en papel. Las eIFUs pueden utilizarse en el caso de Dispositivos Médicos implantables, implantables activos, fijos y software (destinado también a personas no expertas).
7. Información de los operadores económicos (EOs)
La etiqueta suele contener la información del fabricante. Sin embargo, en el caso de fabricantes extranjeros, la información del representante autorizado debe colocarse en las etiquetas comerciales.
8. Advertencias y precauciones
Las advertencias y precauciones deben mencionarse en la etiqueta del dispositivo. La información sobre este aspecto puede ser mínima, y los detalles pueden proporcionarse en las IFU.
Los fabricantes también deben adaptarse a los requisitos de etiquetado específicos de cada país. El requisito de idioma depende del Estado miembro de la UE. Puede afectar en gran medida el etiquetado, las instrucciones de uso (IFUs) y el embalaje del dispositivo en términos de tiempo y costes.
Estos requisitos adicionales pueden aumentar aún más la carga del fabricante con la complejidad existente del proceso de etiquetado. El incumplimiento de los mismos puede resultar muy costoso, implicando retiradas de productos y pasos posteriores para la Acción Correctiva y Preventiva (CAPA).
¿Busca asistencia sobre el etiquetado según el EU MDR? Freyr ofrece servicios integrales en el etiquetado de Dispositivos Médicos. No dude en ponerse en contacto con nuestros expertos reglamentarios ahora en – sales@freyrsolutions.com









