
5 minuti di lettura
Il software per dispositivi medici in Corea del Sud è utilizzato per diagnosticare, trattare e monitorare i pazienti nel moderno sistema sanitario. Comprende sia software integrato nei dispositivi medici sia software autonomo che può essere utilizzato su PC, dispositivi mobili e servizi basati sul web. Il Ministero della Sicurezza Alimentare e Farmaceutica (MFDS) della Corea del Sud è responsabile della regolamentazione del software per dispositivi medici e della garanzia della sua sicurezza ed efficacia. Il 5 luglio 2023, l'MFDS ha stabilito i criteri per l'approvazione e l'ispezione del software per dispositivi medici; queste normative forniscono una struttura che i richiedenti possono seguire quando presentano software per l'approvazione o la revisione.
I regolamenti affrontano una varietà di argomenti, inclusi l'ambito di applicazione, i requisiti di documentazione tecnica e i rapporti di verifica della conformità. Esistono standard e linee guida internazionali che si applicano al software per dispositivi medici, oltre alle linee guida MFDS, come lo standard 62304 della Commissione Elettrotecnica Internazionale (IEC) per i processi del ciclo di vita del software e la guida della Food and Drug Administration degli Stati Uniti (US FDA) sulle applicazioni mediche mobili.
Piano di sviluppo del software e Analisi dei requisiti
- Il Piano di Sviluppo del Software delinea l'approccio generale allo sviluppo del software, incluse specifiche, metodi e strumenti di sviluppo. Copre anche la verifica, la gestione del rischio dei dispositivi medici, la gestione della configurazione e la documentazione.
- Analisi dei requisiti stabilisce i requisiti del software per dispositivi medici, incluse le misure di controllo del rischio e i metodi di verifica. Pianificando e analizzando attentamente il processo di sviluppo del software, gli sviluppatori possono garantire che il software risultante soddisfi gli standard necessari di sicurezza ed efficacia.
- Il Rapporto di Verifica della Conformità del Software include una sintesi del piano di sviluppo del software, il numero di controllo del documento del produttore e una panoramica dell'analisi dei requisiti. Aderendo a queste linee guida, il software per dispositivi medici può essere sviluppato con fiducia, sapendo che è stato sottoposto a test rigorosi e soddisfa gli standard necessari di sicurezza ed efficacia.
Verifica e Validazione del Software per Dispositivi Medici
- La verifica del software per dispositivi medici garantisce che il software soddisfi i requisiti specificati.
- La convalida del software per dispositivi medici garantisce che il software soddisfi le esigenze dell'utente e gli usi previsti.
- Il Rapporto di verifica e convalida del software per dispositivi medici descrive il processo di verifica e convalida, inclusi il nome del prodotto, la revisione e i nomi delle persone che hanno esaminato e approvato il rapporto. Il rapporto può variare a seconda delle caratteristiche del software, ma dovrebbe includere una descrizione del software, i metodi di verifica e convalida utilizzati e i risultati dei test.
Ambiente operativo e Software di Origine Sconosciuta (SOUP)
- Se il software dipende da hardware specifico, come il software integrato, il documento tecnico dovrebbe descrivere le specifiche hardware.
- Tuttavia, se il software è autonomo e sviluppato per funzionare su hardware generico, l'ambiente operativo deve essere descritto nella documentazione di base. Ciò include le specifiche minime raccomandate, come Microsoft Windows 10 o superiore.
- Inoltre, se il software del dispositivo medico include Software Commerciale di Provenienza Sconosciuta (SOUP), deve essere creato un ambiente operativo per garantirne il corretto funzionamento. Descrivendo attentamente l'ambiente operativo e gestendo qualsiasi SOUP, gli sviluppatori possono garantire che il loro software per dispositivi medici sia sicuro ed efficace per l'uso previsto.
Gestione del Rischio e Requisiti di Documentazione per Dispositivi Medici
- Il processo di gestione del rischio del software come dispositivo medico include l'identificazione di situazioni pericolose, la definizione di misure di controllo del rischio, la verifica di tali misure e la gestione delle modifiche al software.
- Il documento MFDS-RM sulla gestione del rischio software fornisce informazioni sulla gestione del rischio software.
- Inoltre, i requisiti di documentazione sono essenziali per garantire che il software soddisfi gli standard necessari di sicurezza ed efficacia.
- Il piano di sviluppo del software, l'analisi dei requisiti del software per dispositivi medici e i rapporti di verifica e convalida del software devono essere inclusi nella documentazione.
- Il Rapporto di Verifica della Conformità del Software delinea i requisiti di documentazione; include anche una panoramica dei documenti applicabili e il numero di controllo del documento del produttore.
Figura 1: Processo di gestione del rischio dei dispositivi medici
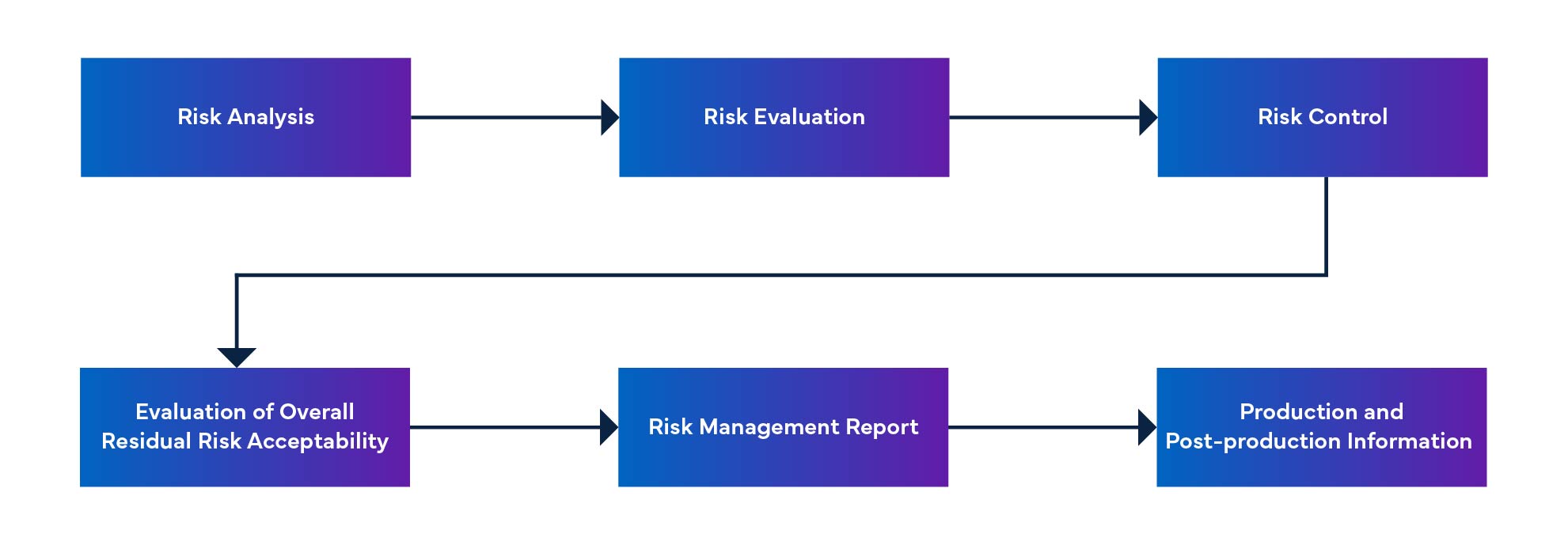
Anomalie irrisolte e azioni correttive per il software SaMD
- Il documento MFDS-PR (Risoluzione dei Problemi Software) delinea il processo di risoluzione dei problemi software, che include la segnalazione, l'analisi, l'implementazione e la verifica dei problemi.
- Il documento include anche un elenco di problemi irrisolti, bug, difetti e anomalie, nonché una valutazione del rischio residuo per il sistema software.
- Le azioni correttive intraprese per affrontare questi problemi devono essere documentate nel piano di manutenzione del software, che è stabilito in base al processo di manutenzione del software.
- Il documento di manutenzione MFDS fornisce informazioni sulla manutenzione e risoluzione dei problemi del software SaMD.
Revisione dei documenti tecnici e requisiti di sottomissione per il software SaMD
I principali documenti di revisione durante il processo di revisione sono i dati sulle prestazioni, il rapporto di conferma di conformità e i dati di verifica e convalida del software del dispositivo medico, la specifica di progettazione del software (SDS), la dichiarazione dei requisiti del software del dispositivo medico (SRS) e i rapporti di verifica e convalida. Il rapporto di conferma di conformità e il rapporto di verifica e convalida del software del dispositivo medico devono essere presentati.
Gestione del Rischio del Software per Dispositivi Medici
- Identificazione dei potenziali pericoli associati al software e al suo utilizzo.
- Valutazione della gravità dei rischi associati a questi pericoli.
- Implementazione di misure di controllo del rischio per minimizzare la probabilità di danno.
- Monitoraggio e revisione dell'efficacia di queste misure di controllo del rischio.
- Documentazione di tutte le attività e decisioni relative alla gestione del rischio dei dispositivi medici.
In un sistema software, gli elementi software vengono suddivisi in parti più piccole, inclusi elementi software dettagliati. Quando un elemento non può essere ulteriormente suddiviso, viene chiamato unità. Il sistema consente la scomposizione a livello di unità, contribuendo a determinare il livello di sicurezza per ogni elemento software. Mettendo insieme questi elementi software, siamo in grado di determinare il livello di sicurezza per l'intero sistema software.
Figura 2: Disassemblaggio e integrazione del software per dispositivi medici
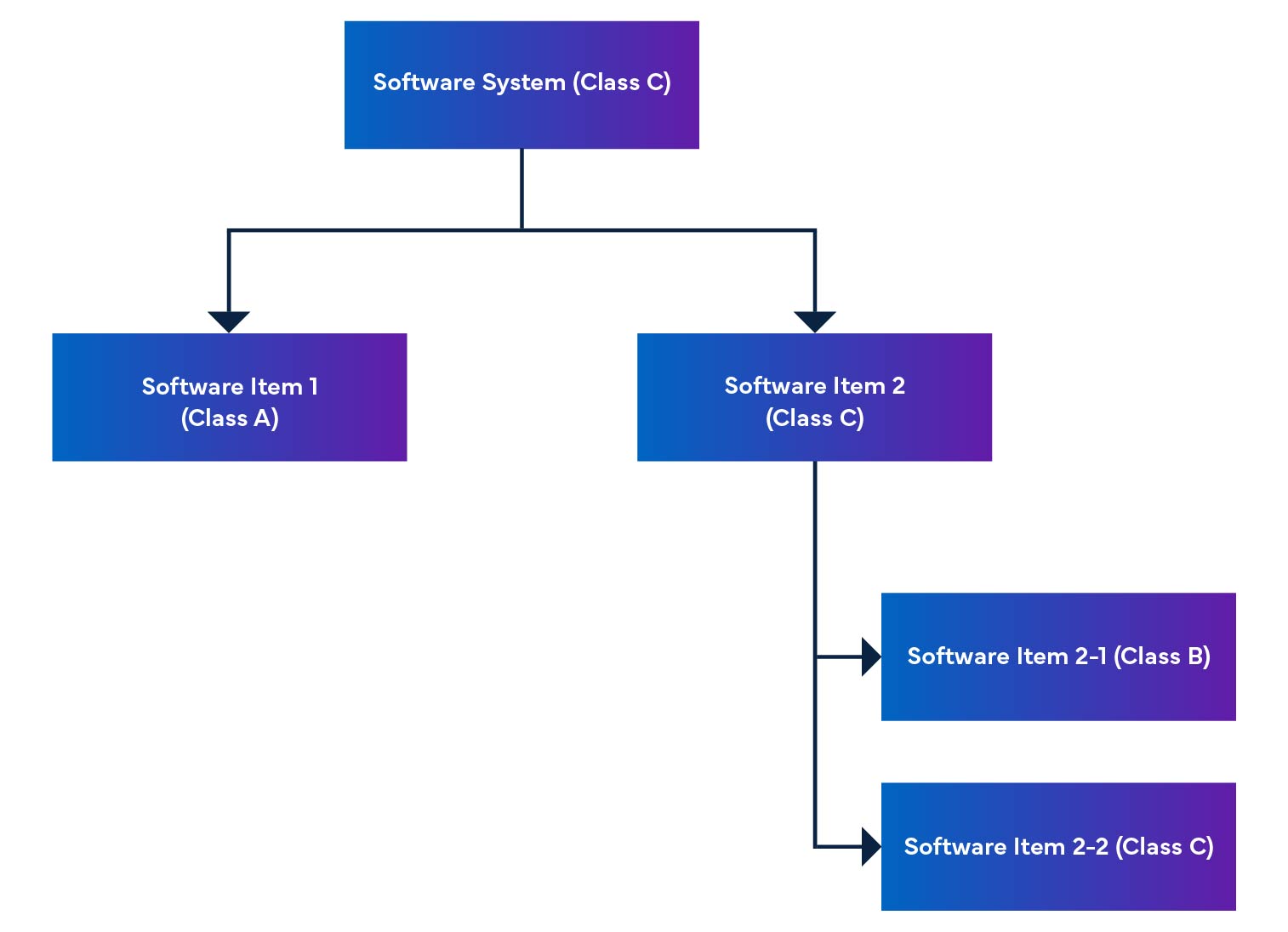
Il regolamento menziona anche la classificazione di sicurezza del software, che è una classificazione per identificare i rischi del software SaMD (vedere Tabella 1).
Tabella 1: Definizione del rating di sicurezza
| Valutazione | Definizione della Classe di Sicurezza del Software per Dispositivi Medici |
| Classe A | Nessuna possibilità di lesioni o danni fisici. |
| Classe B | Sono probabili lesioni meno gravi (lesioni lievi). |
| Classe C | Possibilità di lesioni gravi o morte. |
Gestione della configurazione del software
- Mantenere una documentazione accurata e aggiornata per tutte le versioni, le modifiche e gli aggiornamenti del software.
- Garantire che tutta la documentazione sia correttamente esaminata e approvata.
- Implementazione di procedure per la gestione delle modifiche alla configurazione del software.
- Documentazione di tutte le attività e decisioni relative alla gestione della configurazione del software.
Manutenzione del software
- Testare e monitorare regolarmente il software per assicurarsi che rimanga sicuro ed efficace per l'uso previsto.
- Implementazione di procedure per affrontare eventuali problemi che possano sorgere, inclusi correzioni di bug e aggiornamenti software.
- Documentazione di tutte le attività e decisioni relative alla manutenzione del software.
Risoluzione dei Problemi
- Identificazione della causa principale del problema.
- Implementazione di azioni correttive per affrontare il problema.
- Documentare l'intero processo di risoluzione dei problemi per riferimento futuro.
Seguendo le linee guida di cui sopra, gli sviluppatori possono garantire che eventuali problemi con il software dei loro dispositivi medici siano adeguatamente affrontati e documentati, e che il software soddisfi i requisiti necessari per l'approvazione o l'esame.
Se sei un produttore di dispositivi medici che mira alla conformità con gli standard software per dispositivi medici della Corea del Sud, gli esperti normativi di Freyr possono guidarti attraverso il complesso panorama normativo del paese. Garantiremo che i tuoi dispositivi siano allineati con le più recenti normative sui dispositivi medici della Corea del Sud per una conformità senza intoppi. Contattaci per saperne di più!









