5 min de lectura
Presentaciones programadas para el 24 de septiembre de 2016.
Ahora que se ha implementado la segunda fase del cumplimiento de UDI para los Dispositivos Médicos de Clase III I/LS/LS, muchos fabricantes de dispositivos, especialmente los de tipo Clase II, se preguntan cómo pueden prepararse mejor para la fecha límite del 24 de septiembre de 2016 para la presentación de datos de dispositivos de Clase II. Para anticiparles, en Freyr hemos identificado algunos de los requisitos previos que deberían considerar para preparar los dispositivos de Clase II para el cumplimiento del mandato UDI de la FDA.
La nueva regulación exige que todos los Dispositivos Médicos de clase II sean etiquetados y empaquetados con un identificador único de dispositivo (UDI) y se registren en la Base de Datos Global de Identificación Única de Dispositivos (GUDID) de la FDA. Dada la volatilidad de los requisitos de cumplimiento, junto con plazos de presentación más cortos, el desafío para los fabricantes de dispositivos es conocer a fondo los procesos de cumplimiento. Al mismo tiempo, deben asegurarse de que no se omita ninguno de los atributos clave del dispositivo al recopilar los datos dispersos del dispositivo de diferentes sistemas y conciliarlos en hojas de cálculo para crear informes de cumplimiento.
Para ayudar a los fabricantes de Dispositivos Médicos a navegar fácilmente a través de este proceso de cumplimiento complejo y crítico en el tiempo, sin errores, Freyr ha compilado los siguientes requisitos previos a seguir.
Determinar la Fecha de Cumplimiento de UDI: Desde que la FDA emitió su norma final, algunas de las fechas de cumplimiento de los dispositivos han sido modificadas y extendidas. Para planificar meticulosamente las estrategias y procesos de cumplimiento y evitar enmiendas apresuradas de última hora, los etiquetadores deberán determinar la fecha de cumplimiento precisa.
Fecha de cumplimiento para Dispositivos de Clase II Requisitos de cumplimiento 24 de septiembre de 2016Los dispositivos de Clase III que deben etiquetarse con un UDI deben llevar un UDI como marcado permanente en el propio dispositivo si este está destinado a ser utilizado más de una vez y a ser reprocesado antes de cada uso. Las etiquetas y los envases de los dispositivos médicos de clase II deben llevar un UDI. Las fechas en las etiquetas de estos dispositivos deben formatearse según lo requerido El software independiente de Clase II debe proporcionar su UDI según sea necesario Los datos para dispositivos de clase II que deben llevar una etiqueta con un UDI deben enviarse a la base de datos GUDID Para la mayoría de los dispositivos, la fecha de cumplimiento para el marcado directo es diferente a la de otros requisitos. Según la categoría de su producto, ya sea que esté destinado a ser reutilizado o reprocesado, determine la fecha de cumplimiento del marcado directo UDI como se muestra aquí:
Fechas de Cumplimiento del Marcado Directo Categoría de Dispositivo – Reutilizado y Reprocesado 24 de septiembre de 2015 Dispositivos de soporte vital y de mantenimiento de la vida, independientemente de la clase del dispositivo 24 de septiembre de 2016 Dispositivos de Clase III y dispositivos autorizados en virtud de la Ley de Servicios de Salud Pública 24 de septiembre de 2018 Productos sanitarios de clase II 24 de septiembre de 2020 Dispositivos de Clase I y dispositivos no clasificados Evaluar la necesidad de Marcado Directo del número UDI: Todos los Dispositivos Médicos que se utilizan más de una vez o que deben reprocesarse antes de cada uso deben llevar un marcado directo del UDI. La excepción son los dispositivos implantables que no requieren marcado directo según la norma UDI. Los dispositivos de un solo uso, incluso si se reprocesan, tampoco están obligados a llevar un UDI permanente – 21 CFR 801.45(d)(3). Por lo tanto, evalúe la necesidad de Marcado Directo basándose en la categoría de Dispositivos Médicos que fabrica.
- Plan para un cumplimiento integral: Revise los requisitos de la FDA que deben cumplir sus productos específicos. Realice un análisis exhaustivo de las deficiencias para identificar las carencias relacionadas con los datos o la tecnología y abordar algunos de los principales desafíos en el proceso de cumplir los estrictos plazos de la FDA. Algunos de los desafíos podrían ser la obtención de la información DI o PI, y el manejo de grandes volúmenes de datos no estructurados de diversas fuentes, etc. En lugar de trabajar hasta altas horas de la noche para conciliar todos los datos de Dispositivos Médicos en el último momento, planifique un cumplimiento integral con antelación mediante sistemas y herramientas validados que apoyen la integración, la calidad y la gestión de datos.
![]()
Obtener número DI y la membresía de la agencia: El UDI se compone de un Identificador de Dispositivo (DI – número único basado en la versión o modelo del dispositivo) y un Identificador de Producto (PI – incluye número de lote, número de serie o fecha de caducidad). La parte DI del UDI servirá como clave principal para buscar información sobre el dispositivo en el GUDID. Para asignar el DI, la FDA ha acreditado tres Agencias Emisoras – GS1, HIBCC e ICCBBA. En este escenario, los etiquetadores deben obtener la membresía de una de las agencias para obtener el número DI que debe introducirse en el GUDID de la FDA.
![]()
Identificador Atributos Agencias emisoras UDI DI (Identificador de dispositivo – Datos estáticos)
Debe sincronizarse con GUDIDNúmero único de
Fabricante.
Fabricante del dispositivo
Modelo del dispositivo
GSI
HIBCC
ICCBBAPI (Identificador de Producto – Datos Dinámicos)
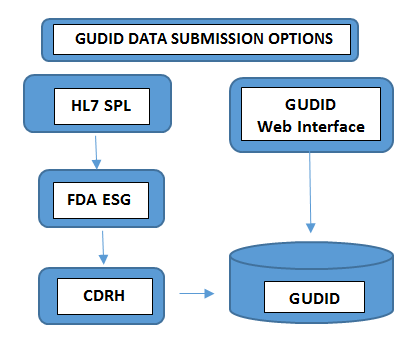
Obligatorio en todos los niveles de envasadoNúmero de lote, número de serie, fecha de fabricación, fecha de caducidad, - Enviar los Datos: La forma en que se envían los datos a GUDID varía según el volumen de carteras de productos que se gestionen. Los fabricantes de Dispositivos Médicos con un número mínimo de dispositivos optan por enviar la información UDI manualmente a través de la interfaz web gratuita de GUDID de la FDA. En este caso, solo se puede enviar un registro DI a la vez a través de una interfaz web segura de GUDID. En el otro caso, los fabricantes con un mayor número de carteras de productos eligen la opción de envío HL7 SPL para recopilar datos electrónicamente y convertir los datos consolidados a formato SPL antes de enviarlos a la Pasarela de Envío Electrónico (ESG) de la FDA, utilizando el número DUNS. Tenga en cuenta que la cuenta GUDID no se define por el tipo de envío. La cuenta sirve para identificarle a usted, el etiquetador, y permitir el envío de datos de dispositivos a través de ambas opciones.
![]()
- Configurar una Cuenta GUDID: Un etiquetador / fabricante de dispositivos requiere una o más cuentas GUDID según el número de roles a asignar; por nombrar algunos, coordinador GUDID, usuario de entrada de datos, etc. Pero para autorizar cada rol para la entrada de datos, el fabricante necesita una aprobación de la FDA antes de la creación de la cuenta. El proceso de creación de una cuenta GUDID adecuada implica enviar una solicitud por correo electrónico a la FDA, tras lo cual el solicitante, usted, recibirá un documento de solicitud de cuenta para rellenar. Una vez que envíe el documento rellenado a la FDA por correo electrónico, la agencia revisará el formulario y luego enviará un correo electrónico con la información de inicio de sesión de la cuenta GUDID.


La implementación de UDI es un proceso complejo y que consume mucho tiempo. Durante el proceso, al cumplir con los requisitos UDI de la FDA, los fabricantes de Dispositivos Médicos se enfrentan a muchos desafíos relacionados con la gestión de datos, la integración de datos y la presentación de datos. Con la fecha límite de cumplimiento para los dispositivos de Clase II a solo un año, Freyr recomienda que las empresas comiencen a trabajar en ello ahora mismo.
Para guiar a su organización a través de este complejo proceso de cumplimiento, Freyr ofrece lo mejor de ambos mundos: una solución de software UDI a la carta, totalmente configurable, Freyr IDENTITY, así como un Centro de Excelencia (CoE) que ofrece servicios UDI de primera clase, rentables y personalizables, diseñados para sus requisitos únicos y exigentes.









