
3 min de lectura
La 510(k) es una presentación previa a la comercialización realizada ante la FDA para demostrar que el dispositivo que se va a comercializar es tan seguro y eficaz, es decir, sustancialmente equivalente a un dispositivo comercializado legalmente (predicado). Los dispositivos de riesgo moderado deben presentar una notificación 510(k), que incluye una minoría de dispositivos de Clase I y III y una mayoría de dispositivos de Clase II.
Existen tres (03) tipos de programas 510(k): Tradicional, Abreviado y Especial. La vía de seguridad y rendimiento se introdujo en 2019 y se basó en el programa abreviado. El programa eSTAR, introducido en 2020, permite la presentación completa de un Dispositivo Médico a través de un formulario PDF interactivo.
¿Quién necesita una certificación 510(k)?
El 510(k) es esencialmente el nombre del proceso/vía que los fabricantes de Dispositivos Médicos que tienen la intención de comercializar sus dispositivos de riesgo moderado a alto en los US siguen para demostrar que el producto a comercializar es tan seguro y eficaz como un dispositivo comercializado legalmente.
A continuación se detalla el proceso paso a paso para obtener una autorización 510(k).
Paso 1- Identificación del Código de Clase de Dispositivo, Tipo de Presentación y Dispositivo de Referencia
- Identificar el código de producto y el número de reglamentación - Para determinar los requisitos de prueba 510(k), es necesario primero identificar el código de producto y el número de reglamentación. Se puede iniciar una búsqueda en la base de datos de la FDA para encontrar el número de reglamentación de 7 dígitos cuya identificación coincida con el uso previsto del dispositivo en cuestión.
- El código de producto de la FDA consta de tres (03) letras. Con este código se puede encontrar información sobre la clasificación del producto, la descripción de la reglamentación y los requisitos de las BPF.
- Selección del tipo de presentación: Un solicitante puede elegir uno de los tres (03) tipos de presentación mencionados anteriormente. 510(k) tradicional es para presentaciones por primera vez, 510(k) especial es para fabricantes de Dispositivos Médicos que desean presentar cambios a un dispositivo existente y se puede elegir 510(k) abreviado cuando el dispositivo cumple con los estándares de consenso voluntario establecidos. En el caso de un 510(k) abreviado, el solicitante debe basarse en los documentos de orientación de la FDA.
- Identificación del Dispositivo Médico de referencia: Un fabricante de Dispositivos Médicos debe demostrar que el dispositivo que pretende comercializar tiene el mismo uso previsto y las mismas características técnicas que el dispositivo comercializado legalmente, también conocido como dispositivo de referencia. Si existen diferencias en las características técnicas, el solicitante debe demostrar que no hay problemas de seguridad y eficacia asociados a dicha diferencia.
Paso 2- Preparación del Expediente 510(k)
El siguiente paso es preparar el expediente 510(k), la guía y la información, que está disponible en el sitio web de la FDA. Incluye la lista de verificación de aceptación para los tres (03) tipos de programas 510(k) y un micrositio titulado Contenido para 510(k), que incluye información sobre declaraciones de indicaciones de uso, comparación de equivalencia sustancial y etiquetado propuesto, entre otra información útil.
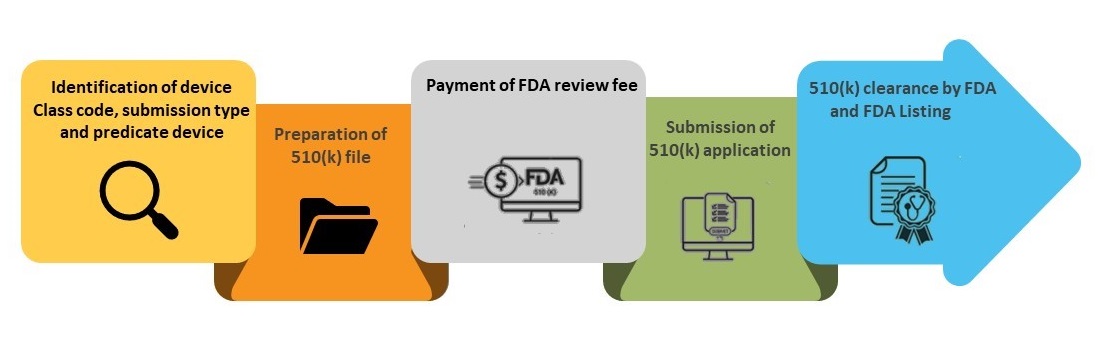
Pasos del proceso de presentación 510(k)
Paso 3 - Pago de la tarifa de revisión de la FDA
Todos los tipos de solicitudes 510(k) están sujetos a la tarifa de usuario. Para el año fiscal 2023, la tarifa estándar para 510(k) es de $19,870. Para las empresas certificadas por el Centro de Diagnóstico y Salud Radiológica (CDRH), también conocidas como pequeñas empresas, la tarifa es de $4,967. La tarifa está sujeta a cambios en el próximo año fiscal.
Paso 4- Presentación de la Solicitud 510(k)
El solicitante puede enviar una copia electrónica (eCopy) o una presentación previa a la comercialización de la Plantilla y Recurso de Presentación Electrónica (eSTAR) a través del portal del CDRH.
A partir del 1 de octubre de 2023, todas las presentaciones 510(k), a menos que estén exentas según la orientación final, deben presentarse como presentaciones electrónicas utilizando eSTAR.
Después de presentar el 510(k), se asigna un número de control único, conocido como “número 510(k)” o “número K”. La FDA realiza dos comprobaciones de verificación, una para verificar si se ha pagado la tarifa de usuario correspondiente y la segunda para verificar si se proporcionó una eCopy o eSTAR válida.
- Para el día 07, la FDA envía una carta de acuse de recibo si se ha pagado la tarifa de usuario correspondiente y se ha proporcionado una eCopy o eSTAR válida. Si no, la FDA envía una carta de retención por problemas sin resolver.
- Para el día 15, la FDA realiza una revisión de aceptación. La FDA informa al solicitante si el 510(k) es aceptado para una revisión sustantiva o si se le aplica una retención por rechazo de aceptación (RTA).
- Para el día 60, la FDA realiza una revisión sustantiva. La FDA se comunica a través de una
interacción sustantiva para informar que la FDA procederá con una revisión interactiva, o bien el 510(k) será puesto en espera y se solicitará información adicional.
Paso 5 - Aprobación de la FDA y registro en la base de datos FDA 510(k)
El objetivo de la FDA es anunciar su decisión sobre las Enmiendas a las Tasas para Usuarios de Dispositivos Médicos (MDUFA) en 90 días FDA. Los días FDA son los días calendario entre la fecha en que se recibió el 510(k) y la fecha de una decisión MDUFA, excluyendo los días en que la presentación estuvo en espera de una solicitud de información adicional. Las decisiones MDUFA para las presentaciones 510(k) incluyen hallazgos de sustancialmente equivalente (SE) o no sustancialmente equivalente (NSE).
Cuando se toma una decisión, la FDA envía una carta de decisión al solicitante por correo electrónico. Una solicitud 510(k) que recibe una carta de decisión SE se considera “aprobada.” Luego se incluye en la base de datos 510(k) junto con las indicaciones de uso del dispositivo médico y el resumen 510(k) o la declaración 510(k) como anexos.
Se puede concluir que una planificación y ejecución cuidadosas, a través de una documentación exhaustiva y una comprensión profunda del entorno reglamentario, son cruciales para una presentación 510(k) exitosa a la FDA.
Para obtener ayuda con el proceso de presentación 510(k) de su Dispositivo Médico, puede escribirnos a sales@freyrsoltions.com, o programar una llamada con nuestros expertos, quienes pueden ayudarle a navegar por los procedimientos. Manténgase informado. Manténgase conforme.









