2 min di lettura
Un “dispositivo precedente” è un dispositivo medico che è stato precedentemente approvato dalla US Food and Drug Administration (US FDA) ed è già sul mercato, che serve come punto di riferimento per i nuovi dispositivi medici che cercano l'approvazione attraverso il percorso di autorizzazione 510(k) della FDA.
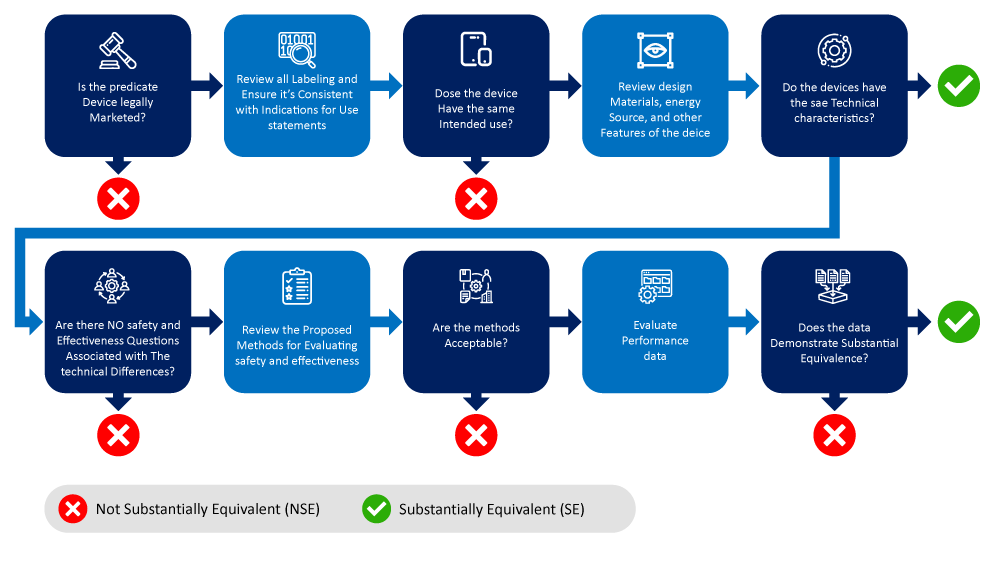
Il dispositivo in questione deve dimostrare di essere almeno altrettanto sicuro ed efficace del dispositivo di riferimento in termini di uso previsto e caratteristiche tecnologiche. Questo confronto è noto come determinazione di "equivalenza sostanziale".
Un nuovo dispositivo non deve essere identico al dispositivo di riferimento per essere sostanzialmente equivalente ad esso.
Come identificare un dispositivo di riferimento?
Il database della FDA fornisce un codice prodotto di tre lettere per ogni classificazione di dispositivo. Il database FDA 510(k) contiene informazioni su tutti i dispositivi autorizzati tramite il processo 510(k). Una volta ottenuto il codice prodotto di tre lettere, è possibile ottenere un elenco di ogni prodotto, ogni azienda e il nome commerciale di ogni concorrente o potenziale concorrente che si desidera esaminare. È quindi possibile effettuare un'analisi e un confronto approfonditi per individuare un dispositivo di riferimento.
Di seguito è riportato un diagramma di flusso che illustra il processo di identificazione e selezione di un dispositivo di riferimento.
Fattori da considerare nel determinare il/i dispositivo/i di riferimento.
- Uso previsto: L'uso previsto dopo il dispositivo di riferimento dovrebbe essere simile a quello del nuovo dispositivo. Ad esempio, se il nuovo dispositivo è destinato al monitoraggio cardiaco, anche il dispositivo di riferimento dovrebbe essere un dispositivo per il monitoraggio cardiaco.
- Caratteristiche tecnologiche: Il dispositivo di riferimento dovrebbe essere identico al nuovo dispositivo in termini di caratteristiche tecnologiche. Si considerino, ad esempio, il design, i materiali utilizzati e il metodo di funzionamento che dovrebbero essere simili.
- Biocompatibilità: Le valutazioni di biocompatibilità di un dispositivo medico o di un componente non dovrebbero limitarsi alle materie prime utilizzate nel dispositivo e nel processo di fabbricazione; dovrebbero essere considerate anche sostanze chimiche aggiuntive. Questo fattore, tuttavia, non si applica agli IVD.
- Tecnologia più recente: Il dispositivo di riferimento non dovrebbe essere obsoleto e dovrebbe rappresentare la tecnologia medica più recente.
Il dispositivo di riferimento è un fattore chiave per determinare se un nuovo dispositivo medico può essere immesso sul mercato attraverso il percorso 510(k). La scelta del dispositivo di riferimento sbagliato potrebbe comportare un processo di approvazione normativa più costoso e lungo, mentre la scelta del dispositivo di riferimento corretto può contribuire a ridurre i costi e i tempi necessari per immettere un nuovo dispositivo medico sul mercato. Se il dispositivo di riferimento non è adatto, ciò potrebbe comportare ritardi e spese aggiuntive.
Per assistenza con il processo di presentazione 510(k) del tuo dispositivo medico, fissa una chiamata con gli esperti normativi di Freyr, che possono aiutarti a orientarti nelle procedure. Rimani informato. Rimani conforme.









