
3 min di lettura
Il Regolamento sui dispositivi medici (MDR) dell'Unione Europea (UE) è da tempo al centro dell'attenzione. L'MDR ha sostituito la Direttiva sui dispositivi medici (MDD) e la Direttiva sui dispositivi impiantabili attivi (AIMDD). Inizialmente, l'intera transizione doveva essere pienamente in vigore entro maggio 2020; tuttavia, a causa dell'emergere della pandemia di COVID-19, l'implementazione è stata posticipata al 26 maggio 2021. In questa tempistica, entro il 26 maggio 2024, tutti i certificati MDD diventeranno nulli e i produttori di dispositivi saranno tenuti a conformarsi all'EU MDR. Inoltre, i dispositivi MDD legalmente immessi sul mercato ai sensi delle Direttive 90/385/CEE e 93/42/CEE prima del 26 maggio 2020, e i dispositivi immessi sul mercato a partire dal 26 maggio 2020 in virtù di un certificato, continueranno ad essere disponibili sul mercato fino al 27 maggio 2025. Le tempistiche sono illustrate di seguito –
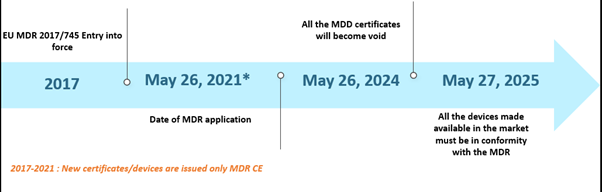
Tempistiche degli scenari passati dell'EU MDR
Tuttavia, la capacità limitata degli Organismi Notificati (NB) e l'impreparazione dei produttori hanno posto alcune sfide nell'implementazione dell'EU MDR secondo la tempistica stabilita. A ottobre 2022, in totale, ci sono trentotto Organismi Notificati (NB), e questi NB hanno ricevuto circa 8120 domande per la certificazione EU MDR, di cui 1990 certificati sono stati rilasciati. Secondo le loro stime con la tempistica iniziale, solo 7000 certificati potevano essere elaborati, e questo ha ulteriormente portato all'estensione della tempistica. Inoltre, uno degli altri probabili motivi dell'estensione era garantire la continua disponibilità di dispositivi medici sicuri i cui certificati sono già scaduti o scadranno prima del 26 maggio 2024. Lo scenario attuale per la tempistica estesa è illustrato di seguito-
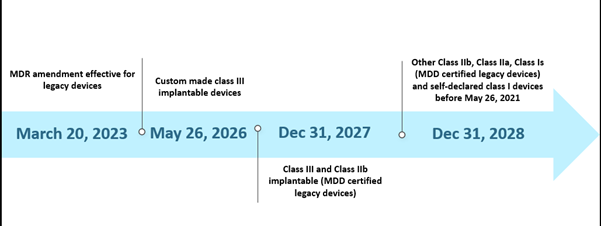
Tempistiche degli scenari passati dell'EU MDR
La nuova estensione è applicabile ai dispositivi preesistenti che soddisfano l'articolo 120 (3e) con una marcatura CE MDD valida o una deroga al 20 marzo 2023, e rimarrà sul mercato insieme ai dispositivi con marcatura CE MDR. Entro il 26 maggio 2024, i produttori di dispositivi preesistenti dovrebbero avere un sistema di gestione della qualità (SGQ) implementato e aver presentato una domanda a un organismo notificato (NB) designato per l'MDR per la valutazione della conformità, ed entro il 26 settembre 2024, i produttori di dispositivi preesistenti dovrebbero avere un accordo con un organismo notificato (NB) designato per l'MDR.
Ora esaminiamo l'impatto che i produttori potrebbero avere con questa estensione.
Opportunità che i produttori hanno con questa estensione:
- Accesso esteso al mercato per i produttori di dispositivi certificati MDD/AIMDD che hanno già intrapreso iniziative di conformità MDR.
- I produttori certificati MDR i cui certificati CE MDD/AIMDD non sono stati revocati sono autorizzati a immettere sul mercato dispositivi legacy fino alla fine del periodo di transizione, oltre ai loro dispositivi conformi al MDR.
- I produttori che hanno una deroga nazionale a partire dal 20 marzo 2023 possono beneficiare del periodo transitorio.
- Il periodo di estensione concede più tempo per una migliore comprensione delle norme e dei regolamenti, il che aiuta a semplificare il processo e a raggiungere la conformità MDR.
Sfide che potrebbero sorgere per i produttori con questa estensione:
- Non c'è alcun vantaggio di mercato per i produttori di dispositivi legacy che non hanno voluto conformarsi al MDR.
- L'estensione dell'MDR può causare il prolungamento dei processi di certificazione e ritardare i lanci di prodotti, il che è una conseguenza diretta dell'arretrato delle revisioni da parte degli NB.
Quali azioni dovrebbero intraprendere i produttori?
- È imperativo che i produttori determinino la classe di rischio MDR del loro dispositivo medico per identificare prontamente la tempistica di transizione appropriata secondo le normative MDR modificate.
- Per garantire la conformità ai regolamenti MDR, è fondamentale identificare e avviare la comunicazione con gli NB designati MDR che possiedono la competenza specifica richiesta per la classificazione del vostro dispositivo medico.
- È fondamentale eseguire una valutazione completa delle lacune per il vostro dispositivo medico certificato ai sensi della MDD/AIMDD, identificare e affrontare eventuali non conformità con i regolamenti MDR e garantire la conformità tempestiva.
È essenziale che i produttori agiscano immediatamente per garantire la conformità con il MDR. La tempistica estesa offre alcune opportunità ai produttori per raggiungere la conformità MDR, ma presenta anche sfide, come processi di certificazione ritardati e il costo della conformità. Per affrontare queste sfide e capitalizzare le opportunità, lasciate che il nostro team di professionisti vi assista nel processo di conformità MDR e assicuri il vostro successo in questo impegnativo ambiente normativo. Prenotate un appuntamento con noi oggi stesso per saperne di più su come possiamo aiutarvi a raggiungere la conformità MDR e a rimanere all'avanguardia. Rimanete informati. Rimanete conformi.









