
3 min di lettura
Il protocollo di validazione è definito come un piano documentato per il test di un dispositivo medico al fine di confermare che il processo di produzione utilizzato per fabbricare il prodotto soddisfa i requisiti specifici dell'utente, tecnici e normativi. Ciò include una revisione delle variabili di processo e delle limitazioni operative e l'analisi dei risultati dei test in condizioni di utilizzo reali.
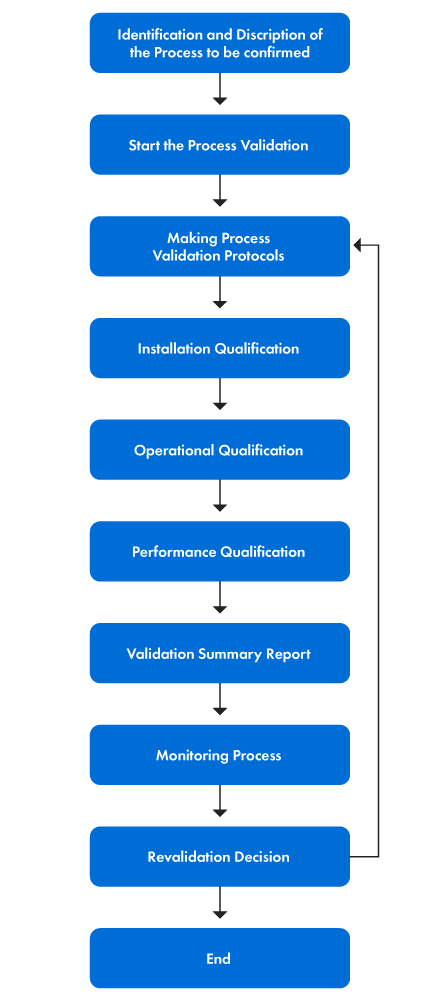
Il processo di convalida comporta diverse azioni concrete. I passaggi sono illustrati come segue:
- Innanzitutto, viene formato il team di convalida e a ciascun membro vengono assegnati ruoli e responsabilità specifici. Lo scopo della convalida del processo è fornire una chiara dichiarazione degli obiettivi di convalida e definire l'ambito delle attività di convalida specificando gli aspetti del dispositivo medico che vengono convalidati. Il team comprende quindi i principi fondamentali del processo per identificare parametri specifici e risultati desiderati.
- In secondo luogo, vengono stabiliti i criteri di valutazione e accettazione, insieme alla selezione dei metodi di prova, degli strumenti e delle tecniche di analisi statistica appropriati. Successivamente, vengono redatti i protocolli di convalida del processo e vengono implementate la qualifica di installazione (IQ), la qualifica operativa (OQ) e la qualifica delle prestazioni (PQ).
- Infine, vengono stabiliti controlli di processo continui e misure di monitoraggio per garantire la convalida continua del processo. Ogni volta che è necessario, viene eseguita una riconvalida per mantenere l'accuratezza e l'efficacia del processo di convalida.
La Figura 1 seguente illustra passo dopo passo il processo di convalida.
Figura 1: Le fasi del processo di convalida
PVP
Data l'ampia gamma di volumi di produzione e le complessità di fabbricazione, esistono numerosi approcci per condurre la convalida del processo. Tuttavia, i regolamenti della Food and Drug Administration degli Stati Uniti (USFDA) e la ISO 13485 forniscono suggerimenti limitati su metodi specifici. Ciononostante, una fonte ampiamente riconosciuta e autorevole per la convalida del processo dei dispositivi medici è un documento guida della Global Harmonization Task Force (GHTF), ora denominata International Medical Device Regulators Forum (IMDRF), pubblicato nel 2004. Rimane il riferimento principale anche sul sito web ufficiale della USFDA.
Secondo il documento guida, viene formato un team di convalida per creare un Piano di Convalida del Processo (PVP) dettagliato. I protocolli di convalida del processo includono uno schema dettagliato su come implementare IQ, OQ, PQ e la riconvalida. Il PVP dovrebbe contenere i seguenti elementi:
- Definire il dispositivo e determinare l'approccio di convalida.
- Identificazione degli elementi che richiedono convalida.
- Svolgimento delle attività presso il sito designato.
- Descrizione dell'ambito della documentazione.
- Creazione di un programma per le attività di convalida.
- Sviluppare un programma generale principale.
- Mantenere un elenco completo e i riferimenti a tutte le validazioni interne ed esterne che sono state eseguite.
Il protocollo di convalida viene redatto prima di condurre le attività di convalida. Dovrebbe essere preparato dal team di convalida e approvato dal dipartimento interessato. Lo scopo di un protocollo di convalida è definire gli script di test che devono essere seguiti per garantire che i processi e le attrezzature siano pronti a fabbricare prodotti dispositivi medici sicuri ed efficaci.
Un rapporto analitico che contiene informazioni insieme alle analisi, spiegazioni e raccomandazioni necessarie, fa parte del protocollo di convalida. Questi registri vengono ulteriormente esaminati per garantire che siano soddisfatti i seguenti due criteri:
- Rispetto degli standard normativi.
- Tutti i registri e i dati generati sono esaminati per verificarne i risultati, l'adeguatezza e la completezza.
La Figura 2 seguente rappresenta il PVP e i vari processi in esso coinvolti.
Figura 2: Il PVP e i suoi requisiti
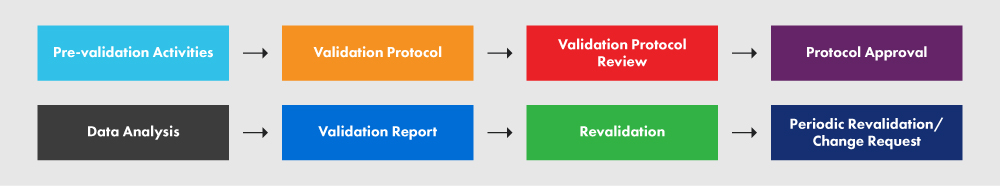
Un protocollo redatto in modo appropriato fornisce chiare linee guida, politiche e procedure da seguire durante la convalida del processo. Esso comprende aspetti quali strutture, attrezzature, metodi e formazione. Il protocollo specifica gli input e i limiti del processo, nonché i passaggi essenziali per la corretta esecuzione del progetto di convalida del processo. Sebbene il seguente schema non includa ogni singolo elemento richiesto nel vostro protocollo, fornisce una panoramica del livello di dettaglio necessario. Raccomandiamo vivamente di seguire il documento guida per una migliore comprensione del processo.
- Frontespizio
- Prodotti da coprire
- Attrezzatura/Processo da convalidare
- Generale
- Obiettivi
- Documenti di riferimento
- Piano di Validazione
- IQ
- OQ
- PQ
- Apparecchiature di misurazione/collaudo e calibrazione
- Manutenzione delle attrezzature
- Riconvalida
- Pagina di Approvazione/Firma del Team di Validazione
La gestione delle operazioni svolge un ruolo cruciale nel mantenere prestazioni ottimali monitorando le misure chiave, rivedendo i metodi e le procedure di lavoro e agendo prontamente quando si presentano problemi. In caso di problemi, potrebbe essere necessario rivalutare un processo parzialmente o anche completamente. Secondo la Sezione 820.75(c) del Regolamento sul Sistema Qualità (QSR) della USFDA, la rivalutazione del processo dovrebbe essere considerata in queste circostanze: “Quando si verificano modifiche o deviazioni di processo, il fabbricante deve esaminare, valutare ed eseguire la rivalutazione, se del caso. Queste attività devono essere documentate.”
I possibili fattori scatenanti per la rivalutazione del processo includono modifiche a specifiche, metodi, procedure, software, progetti, componenti chiave, dimensionamento dei lotti, modifiche di ubicazione, modifiche delle attrezzature e simili. Inoltre, l'implementazione di Azioni Correttive e Preventive (CAPA) può anche fungere da fattore scatenante per la rivalutazione del processo. Le ragioni principali per la rivalutazione sono le seguenti:
- Modifiche apportate al processo.
- Tendenza negativa nella qualità, improvviso deterioramento della qualità o un aumento improvviso dei reclami dei clienti.
- Ampio ampliamento della capacità della linea.
- Modifiche al design.
- Modifiche all'imballaggio del prodotto.
- Trasferimento di un processo a un'altra struttura.
- Modifiche al processo di richiesta.
Per saperne di più sui protocolli di convalida e sulla loro importanza nel campo della produzione di dispositivi medici, consultateci. Rimanete informati! Rimanete conformi!









