3 minuty czytania
Etykietowanie jest integralną częścią marketingu wyrobów medycznych. Etykieta to informacja umieszczona na urządzeniu i/lub opakowaniu w formacie czytelnym dla człowieka. Głównym celem etykietowania jest dostarczenie informacji dotyczących bezpieczeństwa użytkownikom, którymi mogą być pracownicy służby zdrowia, konsumenci lub inne zainteresowane osoby.
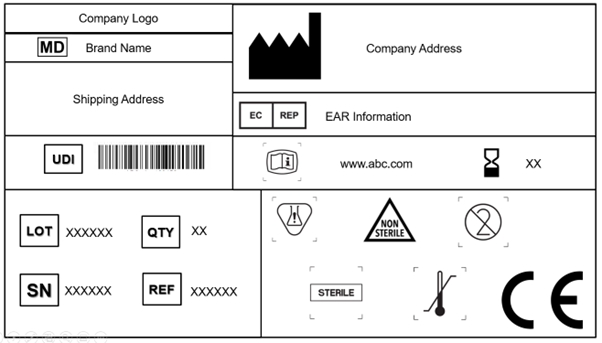
Wszystkie globalne organy regulacyjne mają określone wymagania dotyczące etykietowania. Podobnie, UE szczegółowo określiła wymagania dotyczące etykietowania w Rozdziale III, w Załączniku I do Rozporządzenia UE w sprawie wyrobów medycznych (EU MDR) 2017/745. Najważniejszą rzeczą, na którą należy zwrócić uwagę, jest uwzględnienie wszystkich symboli zawierających wymagane informacje w oznakowaniu wyrobu oraz w towarzyszących mu dokumentach (broszurach, instrukcjach, IFU itp.).
Niektóre z kluczowych kwestii dotyczących etykietowania, które należy wziąć pod uwagę podczas przestrzegania przepisów EU MDR 2017/745, to:
1. Symbologia oznakowania wyrobów medycznych
Każdy producent jest zobowiązany do umieszczenia symbolu wyrobu medycznego, który informuje, że produkt dostarczony na rynek UE jest wyrobem medycznym. Obowiązkowe jest umieszczenie tego symbolu na wyrobie i na wszystkich poziomach opakowania. Ponadto, etykieta powinna zawierać nazwę handlową i oryginalną nazwę wyrobu.
2. Specjalne urządzenia
W przypadku, gdy produkt jest wyrobem specjalnym lub niestandardowym, jego status powinien być wskazany na etykiecie. Na przykład, jeśli produkt jest przeznaczony wyłącznie do badań klinicznych, etykieta powinna to wyraźnie zaznaczać.
W przypadku wyrobów z materiałami chłonnymi lub które mogą lokalnie rozpraszać się w ludzkim ciele, oznakowanie powinno zawierać skład materiału oraz ilościowe szczegóły dotyczące kluczowych składników.
Nawet w przypadku wyrobów jednorazowego użytku i wyrobów sterylnych wymagane jest wyraźne etykietowanie. W przypadku wyrobów poddanych ponownemu przetworzeniu, etykieta powinna zawierać informację o liczbie możliwych ponownych przetworzeń, liczbie dotychczasowych ponownych przetworzeń oraz zastosowanej metodzie sterylizacji.
3. Obecność substancji toksycznych
Deklaracja obecności substancji CMR (rakotwórczych, mutagennych, toksycznych dla rozrodczości) oraz substancji zaburzających gospodarkę hormonalną jest obowiązkowa na etykietach, jeśli stężenie przekracza 0,1% w/w. Lista takich substancji musi być dołączona do wyrobu i/lub opakowania.
Ponadto, etykieta informująca o obecności pochodnych krwi i tkanek (nawet jeśli są zawarte w substancji leczniczej wyrobu złożonego) musi być umieszczona na wyrobach.
4. Zharmonizowane standardy
EU MDR 2017/745 uznaje i akceptuje normę ISO 15223-1: 2021. Dokument określa symbole, które mają być używane w etykietowaniu wyrobów medycznych i ich opakowaniach. Rozdział 3 (23.1,h) Załącznika I do EU MDR określa, że można stosować międzynarodowo uznane symbole, a w przypadku regionów, gdzie symbole te nie są rozpoznawane, ich opis musi być dostarczony w dokumencie wraz z wyrobem.
5. UDI
Artykuły 27, 28, 29 oraz załącznik VI (A, B, C) szczegółowo określają zasady i przepisy dotyczące UDI. Etykieta musi teraz zawierać nośnik UDI [Automatyczna Identyfikacja do Przechwytywania Danych (AIDC) i Interpretacja Czytelna dla Człowieka (HRI) reprezentacja UDI] na wyrobie oraz na wyższych poziomach opakowania. Wyższe opakowanie wyrobu (z wyłączeniem opakowań transportowych) będzie miało własny nośnik UDI.
6. Elektroniczna informacja dla użytkownika (eIFU)
Adres internetowy (URL) w formie eIFU może być również umieszczony na etykiecie wyrobu medycznego wraz z papierowymi IFU. eIFU mogą być stosowane w przypadku implantowalnych, aktywnych implantowalnych, stałych wyrobów medycznych i oprogramowania (przeznaczonego również dla laików).
7. Informacje o podmiotach gospodarczych (EO)
Etykieta zazwyczaj zawiera informacje o producencie. Jednakże, w przypadku producentów zagranicznych, informacje autoryzowanego przedstawiciela powinny być umieszczone na etykietach handlowych.
8. Ostrzeżenia i środki ostrożności
Ostrzeżenia i środki ostrożności muszą być umieszczone na etykiecie urządzenia. Informacje w tym zakresie mogą być ograniczone do minimum, a szczegóły mogą być podane w instrukcji użytkowania (IFU).
Producenci są również zobowiązani do dostosowania się do wymagań dotyczących etykietowania specyficznych dla danego kraju. Wymóg językowy zależy od państwa członkowskiego UE. Może to znacząco wpłynąć na etykietowanie wyrobu, instrukcje użytkowania i opakowanie pod względem czasu i kosztów.
Te dodatkowe wymagania mogą dodatkowo zwiększyć obciążenie producenta przy istniejącej złożoności procesu etykietowania. Niespełnienie ich może być bardzo kosztowne, wiążąc się z wycofaniem produktów i późniejszymi działaniami korygującymi i zapobiegawczymi (CAPA).
Szukasz pomocy w zakresie etykietowania zgodnie z EU MDR? Freyr oferuje kompleksowe usługi w zakresie etykietowania wyrobów medycznych. Zapraszamy do kontaktu z naszymi ekspertami ds. regulacji pod adresem – sales@freyrsolutions.com









