2 min de lectura
Un “dispositivo precedente” es un Dispositivo Médico que ha sido previamente aprobado por la Administración de Alimentos y Medicamentos de US (US FDA) y ya está en el mercado, que sirve como punto de referencia para nuevos Dispositivos Médicos que buscan aprobación a través de la vía de autorización 510(k) de la FDA.
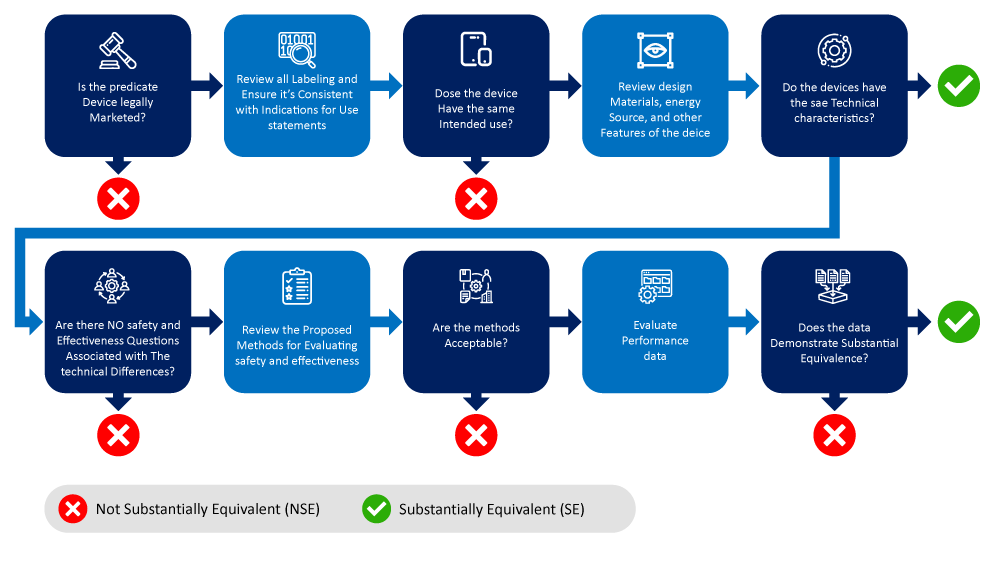
Se debe demostrar que el dispositivo en cuestión es al menos tan seguro y eficaz como el dispositivo de referencia en términos de su uso previsto y características tecnológicas. Esta comparación se conoce como determinación de “equivalencia sustancial”.
Un nuevo dispositivo no necesita ser idéntico al dispositivo de referencia para ser sustancialmente equivalente a este.
¿Cómo identificar un dispositivo de referencia?
La base de datos de la FDA proporciona un código de producto de tres letras para cada clasificación de dispositivo. La base de datos 510(k) de la FDA contiene información sobre todos los dispositivos autorizados a través del proceso 510(k). Una vez que tenga el código de producto de tres letras, puede obtener una lista de cada producto, cada empresa y el nombre comercial de cada competidor o competidor potencial que desee investigar. Luego puede realizar un análisis y una comparación en profundidad para reducir las opciones a un dispositivo de referencia.
A continuación se muestra un diagrama de flujo que describe el proceso de identificación y selección de un dispositivo de referencia.
Factores a considerar al determinar el(los) Dispositivo(s) Predicado(s)
- Uso previsto: El uso previsto después del dispositivo de referencia debe ser similar al del nuevo dispositivo. Por ejemplo, si el nuevo dispositivo está destinado a ser utilizado para la monitorización cardíaca, el dispositivo de referencia también debe ser un dispositivo de monitorización cardíaca.
- Características tecnológicas: El dispositivo precedente debe ser idéntico al nuevo dispositivo en términos de características tecnológicas. Por ejemplo, el diseño, los materiales utilizados y el método de funcionamiento deben ser similares.
- Biocompatibilidad: Las evaluaciones de biocompatibilidad de un Dispositivo Médico o componente no deben limitarse a las materias primas utilizadas en el dispositivo y en el proceso de fabricación; también deben considerarse productos químicos adicionales. Este factor, sin embargo, no se aplica a los IVD.
- Última tecnología: El dispositivo de referencia no debe estar obsoleto y debe representar la última tecnología médica.
El dispositivo de referencia es un factor clave para determinar si un nuevo Dispositivos Médicos puede introducirse en el mercado a través de la vía 510(k). Elegir el dispositivo de referencia incorrecto podría resultar en un proceso de aprobación reglamentaria más costoso y que consume más tiempo, mientras que elegir el dispositivo de referencia adecuado puede ayudar a reducir el costo y el tiempo necesarios para introducir un nuevo Dispositivos Médicos en el mercado. Si el dispositivo de referencia no es adecuado, podría resultar en retrasos y gastos adicionales.
Para obtener ayuda con el proceso de presentación 510(k) de su Dispositivo Médico, programe una llamada con los expertos reglamentarios de Freyr, quienes pueden ayudarle a navegar por los procedimientos. Manténgase informado. Manténgase conforme.









