
3 min de lectura
El Sistema de Gestión de Calidad (SGC) es un componente esencial de la industria de Dispositivos Médicos, garantizando la seguridad, eficacia y el cumplimiento reglamentario de los Dispositivos Médicos a lo largo de todo su ciclo de vida. El SGC se implementa en todas las etapas del ciclo de vida de los Dispositivos Médicos, incluida la fase de diseño y desarrollo, para garantizar que el dispositivo cumpla con los requisitos reglamentarios y del usuario, y que cualquier riesgo potencial sea identificado y abordado.
Figura 1: Etapas del ciclo de vida de un Dispositivo Médico
En este blog, analizaremos la importancia de QMS en la fase de diseño y desarrollo del ciclo de vida de los Dispositivos Médicos.
Fase de Diseño y Desarrollo en el Ciclo de Vida del Dispositivo Médico
La fase de diseño y desarrollo es una de las etapas más críticas en el ciclo de vida de un Dispositivo Médico. Durante esta etapa, se desarrolla el diseño del dispositivo y se crean prototipos, seguidos de pruebas de verificación y validación como parte del ciclo de vida del Dispositivo Médico.
Para asegurar que el Dispositivo Médico cumple con los requisitos reglamentarios, la seguridad, la eficacia y las expectativas del usuario, la implementación de un Sistema de Gestión de Calidad (SGC) es esencial en la etapa de diseño y desarrollo del ciclo de vida de un Dispositivo Médico.
La documentación es crucial durante la fase de diseño y desarrollo de Dispositivos Médicos. El QMS garantiza que toda la documentación relacionada con el diseño y desarrollo esté controlada, gestionada y documentada.
El Expediente de Historial de Diseño (DHF) es un archivo/registro importante, que contiene toda la documentación relacionada con el diseño y desarrollo del dispositivo. El DHF proporciona la evidencia de que el diseño del dispositivo cumple con los requisitos reglamentarios.
El DHF debe contener documentación relacionada con las entradas de diseño, las salidas de diseño, las revisiones de diseño, la verificación de diseño, la validación, los cambios en el diseño y la gestión de riesgos. Obtenga más información sobre el DHF aquí.
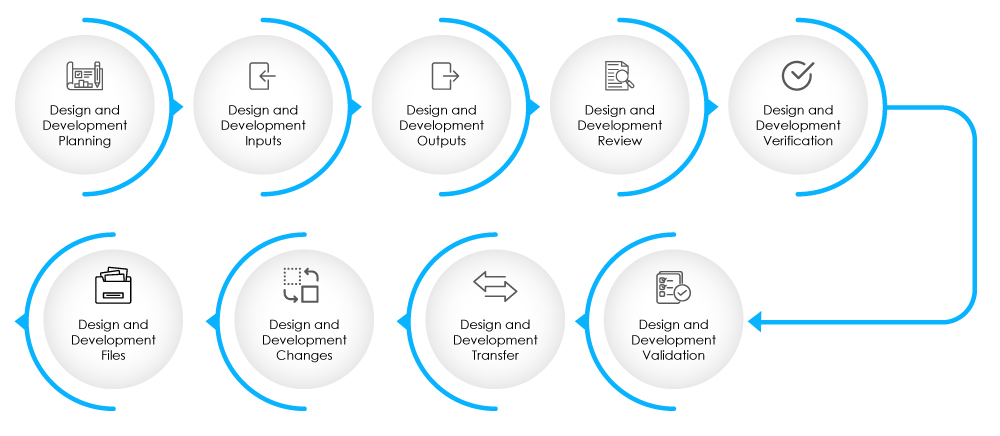
Fig. 2 - Etapas de la fase de diseño y desarrollo
Mejores prácticas para la etapa de diseño y desarrollo
- Establecer un Enfoque Estructurado: Desarrolle un enfoque estructurado para el desarrollo y la gestión del DHF que se adapte a las necesidades específicas de su organización. Este enfoque debe incluir directrices, procedimientos y flujos de trabajo claros para el desarrollo y la gestión del DHF.
- Definir y Documentar los Datos de Entrada del Diseño: Defina y documente claramente los datos de entrada del diseño, incluyendo los requisitos y las especificaciones del dispositivo. Esto puede ayudar a garantizar que el DHF sea exhaustivo y completo.
- Gestionar los cambios de diseño: Implementar un proceso robusto de gestión de cambios que incluya procedimientos para documentar, evaluar y aprobar los cambios de diseño. Esto puede ayudar a garantizar que los cambios se documenten y evalúen correctamente en cuanto a su impacto en la seguridad y eficacia del dispositivo.
- Garantizar la Trazabilidad: Desarrolle una matriz de trazabilidad que vincule los datos de entrada del diseño con los resultados del diseño y asegure que todas las actividades de diseño y desarrollo estén debidamente documentadas y registradas. Esto puede ayudar a garantizar que el DHF sea trazable y que el proceso de toma de decisiones esté bien documentado.
- BEquilibrar Innovación y Cumplimiento: Desarrollar una cultura de innovación asegurando que se cumplan los requisitos de cumplimiento relacionados con el DHF, como los controles de diseño y la gestión de riesgos. Esto se puede lograr desarrollando procedimientos y flujos de trabajo que faciliten la innovación mientras se asegura el cumplimiento de los requisitos reglamentarios.
- Implementar el control de documentos: Implementar procedimientos de control de documentos que garanticen que los documentos del DHF estén debidamente controlados, con control de versiones y accesibles para el personal autorizado. Esto puede ayudar a garantizar que los documentos del DHF estén seguros y que los cambios se documenten y aprueben correctamente.
- Capacitar al equipo: Asegúrese de que el equipo responsable del desarrollo y la gestión del DHF esté debidamente capacitado en los requisitos del DHF y tenga la experiencia técnica para desarrollar el producto. Esto se puede lograr a través de sesiones de capacitación regulares, tutorías y la contratación de profesionales experimentados con las habilidades y la experiencia necesarias.
Al seguir estas mejores prácticas, la industria de Dispositivos Médicos puede garantizar el cumplimiento de los requisitos reglamentarios, promover la seguridad y eficacia de sus productos y mantener su ventaja competitiva en el mercado.
En conclusión, implementar un QMS desde la fase de diseño y desarrollo es fundamental para el éxito en la industria de dispositivos médicos altamente reglamentada. Al mantener registros sistemáticos y cumplir con los requisitos reglamentarios, la industria de dispositivos médicos puede asegurar que está entregando productos de alta calidad y manteniendo la satisfacción del cliente.
En Freyr, ofrecemos servicios de QMS para ayudar a la industria de Dispositivos Médicos a cumplir con los requisitos reglamentarios en todas las etapas del ciclo de vida del Dispositivo Médico. Contáctenos con nuestros expertos en QMS y reglamentarios para saber más.









