5 minuti di lettura
Presentazioni programmate per il 24 settembre 2016.
Ora che la seconda fase della conformità UDI, per i dispositivi medici di Classe III I/LS/LS, è stata implementata, molti produttori di dispositivi, in particolare quelli di tipo Classe II, si stanno chiedendo come possano prepararsi al meglio per la scadenza del 24 settembre 2016 per la sottomissione dei dati dei dispositivi di Classe II. Per dar loro un'indicazione, in Freyr, abbiamo identificato alcuni dei prerequisiti che dovrebbero considerare per allineare i dispositivi di Classe II alla conformità con il mandato UDI della FDA.
La nuova regolamentazione richiede che tutti i dispositivi medici di classe II siano etichettati e confezionati con un identificatore unico del dispositivo (UDI) e inseriti nel database globale di identificazione unica del dispositivo (GUDID) della FDA. Data la volatilità dei requisiti di conformità, unita a tempistiche di presentazione più brevi, la sfida per i produttori di dispositivi è conoscere i dettagli dei processi di conformità. Allo stesso tempo, devono assicurarsi che nessuno degli attributi chiave del dispositivo venga tralasciato mentre raccolgono i dati sparsi dei dispositivi da diversi sistemi e li riconciliano in fogli di calcolo per creare rapporti di conformità.
Al fine di aiutare i produttori di dispositivi a navigare facilmente e senza errori in questo processo di conformità complesso e critico in termini di tempo, Freyr ha compilato i seguenti prerequisiti da seguire.
Determinare la data di conformità UDI: Da quando la FDA ha emesso la sua norma finale, alcune delle date di conformità dei dispositivi sono state modificate ed estese. Per pianificare meticolosamente in anticipo le strategie e i processi di conformità ed evitare modifiche affrettate all'ultimo minuto, gli etichettatori devono determinare la data di conformità precisa.
Data di conformità dei dispositivi di Classe II Requisiti di conformità 24 set 2016I dispositivi di Classe III che devono essere etichettati con un UDI devono riportare un UDI come marcatura permanente sul dispositivo stesso, se il dispositivo è destinato ad essere utilizzato più di una volta e ad essere riprocessato prima di ogni utilizzo. Le etichette e le confezioni dei dispositivi medici di classe II devono recare un UDI. Le date sulle etichette di questi dispositivi devono essere formattate come richiesto Il software autonomo di Classe II deve fornire il proprio UDI come richiesto I dati per i dispositivi di classe II che devono essere etichettati con un UDI devono essere inviati al database GUDID Per la maggior parte dei dispositivi, la data di conformità per la marcatura diretta è diversa dagli altri requisiti. In base alla categoria del prodotto, destinato al riutilizzo o alla rielaborazione, determinare la data di conformità UDI per la marcatura diretta come mostrato qui:
Date di Conformità per la Marcatura Diretta Categoria del dispositivo – Riutilizzato e riprocessato 24 set 2015 Dispositivi salvavita e di supporto vitale, indipendentemente dalla classe del dispositivo 24 set 2016 Dispositivi di Classe III e dispositivi autorizzati ai sensi del Public Health Service Act 24 Set 2018 Dispositivi di classe II 24 Set 2020 Dispositivi di Classe I e dispositivi non classificati Valutare la necessità della marcatura diretta del numero UDI: Tutti i dispositivi medici utilizzati più di una volta o che devono essere riprocessati prima di ogni utilizzo devono avere la marcatura diretta dell'UDI. L'eccezione riguarda i dispositivi impiantabili che non richiedono la marcatura diretta secondo la regola UDI. I dispositivi monouso, anche se riprocessati, non sono tenuti a recare un UDI permanente – 21 CFR 801.45(d)(3). Pertanto, valutare la necessità della marcatura diretta in base alla categoria dei dispositivi medici che si producono.
- Piano per la conformità completa: Esaminare i requisiti della FDA per i vostri prodotti specifici da rispettare. Eseguire un'analisi approfondita delle lacune per individuare le carenze relative ai dati o alla tecnologia al fine di affrontare alcune delle principali sfide nel processo di rispetto delle rigorose tempistiche della FDA. Alcune delle sfide potrebbero essere l'ottenimento delle informazioni DI o PI e la gestione di grandi volumi di dati non strutturati provenienti da fonti disparate, ecc. Invece di lavorare fino a tardi per riconciliare tutti i dati dei dispositivi medici all'ultimo minuto, pianificare in anticipo una conformità completa attraverso sistemi e strumenti validati che supportano l'integrazione dei dati, la qualità dei dati e la gestione dei dati.
![]()
Ottenere il numero DI e l'iscrizione all'agenzia: L'UDI è composto dall'Identificatore del Dispositivo (DI – numero unico basato sulla versione o sul modello del dispositivo) e dall'Identificatore del Prodotto (PI – include il numero di lotto, il numero di serie o la data di scadenza). La parte DI dell'UDI servirà come chiave primaria per ricercare informazioni sul dispositivo nel GUDID. Per assegnare il DI, la FDA ha accreditato tre Agenzie di Emissione – GS1, HIBCC e ICCBBA. In questo scenario, gli etichettatori devono ottenere l'adesione a una delle agenzie per ottenere il numero DI che deve essere inserito nel GUDID della FDA.
![]()
Identificatore Attributi Enti emittenti UDI DI (Identificatore del Dispositivo – Dati Statici)
Richiesto per la sincronizzazione con GUDIDNumero Unico di
Fabbricante
Marca del dispositivo
Modello del dispositivo
GSI
HIBCC
ICCBBAPI (Identificatore di Prodotto – Dati Dinamici)
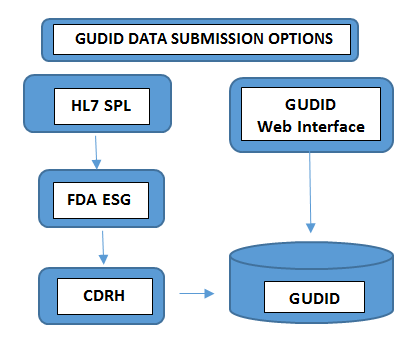
Richiesto su tutti i livelli di imballaggioNumero di lotto, Numero di serie, Data di fabbricazione, Data di scadenza, - Invia i dati: Il modo in cui si inviano i dati a GUDID varia in base al volume di portafogli di prodotti gestiti. I produttori di dispositivi con un numero minimo di dispositivi scelgono di inviare le informazioni UDI manualmente tramite l'interfaccia web gratuita GUDID della FDA. In questo caso, un solo record DI può essere inviato alla volta tramite un'interfaccia web GUDID sicura. Nell'altro caso, i produttori con un numero maggiore di portafogli di prodotti scelgono l'opzione di invio HL7 SPL per raccogliere i dati elettronicamente e convertire i dati consolidati in formato SPL prima di inviarli all'Electronic Submission Gateway (ESG) della FDA, utilizzando il numero DUNS. Si prega di notare che l'account GUDID non è basato sul tipo di invio. L'account serve a identificare l'etichettatore, per consentire l'invio dei dati del dispositivo tramite entrambe le opzioni.
![]()
- Creare un account GUDID: Un etichettatore/produttore di dispositivi richiede uno o più account GUDID in base al numero di ruoli da assegnare; per citarne alcuni, coordinatore GUDID, utente di inserimento dati, ecc. Ma per autorizzare ogni ruolo all'inserimento dei dati, il produttore necessita di un'approvazione dalla FDA prima della creazione dell'account. Il processo di creazione di un account GUDID appropriato prevede l'invio di una richiesta via email alla FDA, dopodiché il richiedente, voi, riceverà un documento di richiesta account da compilare. Una volta inviato il documento compilato alla FDA via email, l'agenzia esaminerà il modulo e invierà un'email con le informazioni di accesso all'account GUDID.


L'implementazione dell'UDI è un processo complesso e che richiede tempo. Nel corso di tale processo, pur soddisfacendo i requisiti UDI della FDA, i produttori di dispositivi medici affrontano molte sfide relative alla gestione dei dati, all'integrazione dei dati e alla presentazione dei dati. Con la scadenza per la conformità dei dispositivi di Classe II a solo un anno di distanza, Freyr raccomanda alle aziende di iniziare a lavorarci subito.
Per guidare la vostra organizzazione attraverso questo complesso processo di conformità, Freyr offre il meglio di entrambi i mondi – una soluzione software UDI su richiesta, completamente configurabile, Freyr IDENTITY, nonché un Centro di Eccellenza (CoE) che offre servizi UDI di prim'ordine, convenienti e personalizzabili, costruiti attorno alle vostre esigenze uniche e impegnative.









