
Visão geral dos serviços de conformidade com o Regulamento IVDR da UE
O Regulamento Europeu sobre Dispositivos Médicos para Diagnóstico In Vitro (IVDR da UE) constitui uma nova base regulamentar para a colocação no mercado europeu de dispositivos médicos para diagnóstico in vitro. O Regulamento IVDR da UE destina-se a substituir a atual diretiva da UE relativa aos dispositivos médicos de diagnóstico in vitro (UE 98/79/CE). Enquanto regulamento europeu, será aplicável em todos os Estados-Membros da UE e nos Estados-Membros da Associação Europeia de Comércio Livre (AELC), sem que seja necessário transpor para o direito nacional dos Estados em causa. Enquanto parceiro regulamentar de renome, a Freyr oferece aos fabricantes serviços de conformidade e consultoria em matéria de IVDR na UE, a fim de garantir a conformidade com os regulamentos relativos ao diagnóstico in vitro e à marcação CE.
Marque uma reunião com os nossos especialistas em IVDR da UE
O Regulamento IVDR - Calendário de transição
Previsto para entrar em vigor a 26 de maio de 2022, o novo regulamento europeu IVDR é considerado uma mudança significativa na supervisão regulamentar dos dispositivos médicos para diagnóstico in vitro. O resumo da transição para o IVDR é o seguinte –
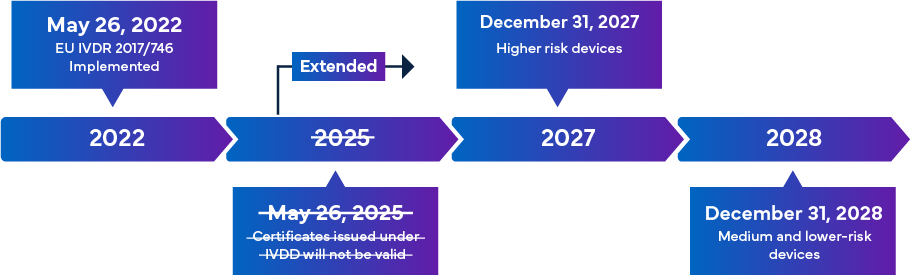
Uma vez plenamente implementadas, as regras de conformidade do IVDR garantem que praticamente todos os dispositivos de diagnóstico in vitro (IVD) que entram no mercado europeu sejam submetidos à avaliação do organismo notificado (ON) do IVDR e à certificação da marcação CE no âmbito do procedimento de aprovação dos IVD. Neste cenário, para além da adaptação à classificação atualizada da IVDR, os fabricantes devem rever imediatamente a documentação técnica essencial para obterem o registo do IVD e a marcação CE. Os requisitos da IVDR variam em função da classe de risco do IVD. Em geral, para a certificação dos IVD, os fabricantes devem realizar as seguintes operações:
- Análise dos dossiês técnicos em conformidade com o Regulamento IVDR (Regulamento (UE) n.º 2017/746)
- Elaboração do relatório de avaliação de desempenho (PER) para todos os dispositivos de diagnóstico in vitro (IVD)
- Acompanhamento do desempenho pós-comercialização (PMPF), em conformidade com o anexo XIII, parte B, do IVDR.
- Relatório de vigilância nos termos do artigo 82.º do IVDR
A Freyr apoia os seus clientes na realização de uma revisão sistemática da literatura científica e ajuda-os a planear e a preparar um PER, de modo a cumprir a regulamentação relativa aos dispositivos de diagnóstico in vitro. A Freyr dispõe de especialistas que prestam serviços de consultoria em matéria de IVDR e apoio à vigilância pós-comercialização (SPM), que constitui parte integrante do sistema de gestão da qualidade do fabricante, bem como ao acompanhamento do desempenho pós-comercialização (SPPM).
Obtenha conselhos de especialistas sobre a conformidade com o Regulamento IVDR da UE
Serviços de conformidade com o Regulamento IVDR da UE: A EXPERIÊNCIA E AS VANTAGENS DE
- Plano de transição para a conformidade com o IVDR
- Análise técnica e identificação de lacunas nos requisitos do IVDR relativos aos GSPR (requisitos gerais de segurança e desempenho)
- Apoiar a elaboração do dossiê técnico em conformidade com os requisitos do IVDR
- Relatórios de validade científica baseados na literatura e/ou em dados internos
- Relatórios sobre o desempenho clínico baseados na literatura e/ou em dados internos
- Dados clínicos ou relatórios de avaliação de desempenho
- Protocolos e relatórios de acompanhamento do desempenho pós-comercialização (SPPM)
- Protocolos e relatórios de vigilância pós-comercialização (SPM)
- Redigir/revisar outros documentos, tais como folhetos informativos/IFU (instruções de utilização), guias de referência rápida (QRI), manuais de utilização/funcionamento, etc.

- Garantir a conformidade com o IVDR, o registo IVD e a marcação CE
- Profundo conhecimento da regulamentação e experiência nas áreas-chave abrangidas pelo IVDR da UE
- Modelo de entrega centrado na gestão de projetos, com vista a garantir o cumprimento do calendário.
- Peritos internos da ON (análise do relatório pelos avaliadores interativos da ON)
- Equipas especializadas com conhecimentos interdisciplinares em áreas de impacto e categorias específicas de dispositivos
- Contribuições interdisciplinares de especialistas em dispositivos médicos para garantir a conformidade com os requisitos
- Conjunto de serviços relacionados com a conformidade, a análise e o planeamento
- Grande experiência na garantia da consistência dos resultados (prazos e qualidade)
