
O que é o QMSR?
FDA é uma abordagem simplificada que se alinha aos requisitos ISO 13485:2016, que constitui uma atualização da anterior estrutura do QSR. Este alinhamento é crucial, pois simplifica a conformidade global para os fabricantes, especialmente aqueles que operam a nível mundial. Esta harmonização permitirá às empresas cumprir os requisitos regulamentares tanto nos US noutros mercados de uma forma muito mais unificada.
A QMSR exige melhorias na gestão de riscos, na conceção dos dispositivos e Vigilância Pós-Comercialização. Esta atualização pode aumentar a complexidade, mas também permite aos fabricantes padronizar os seus procedimentos de qualidade, o que, por sua vez, aumenta a segurança dos dispositivos e melhora a documentação, o que pode ser vital durante USFDA .
Por que razão a transição de QSR para QMSR é crucial?
![]()
![]()
Conformidade com as normas internacionais
Isto ajudará os fabricantes a alinharem-se com o sistema de gestão da qualidade (SGQ) global para os requisitos relativos aos dispositivos médicos, através da transição para o QMSR, o que reduz as redundâncias regulamentares para os fabricantes de dispositivos médicos que operam a nível global.
![]()
Melhoria da qualidade, segurança e eficácia
Esta integração garante que os dispositivos sejam fabricados de acordo com os requisitos globais do Sistema de Gestão da Qualidade. Isto ajuda o fabricante a melhorar a qualidade e a segurança dos dispositivos.
![]()
Conformidade Regulatória
Isto ajuda os fabricantes a adaptarem-se aos novos requisitos e reduz o risco de incumprimento quando o regulamento entrar em vigor a 2 de fevereiro de 2026.



Principais alterações na transição de QSR para QMSR:
![]()
Harmonização com a terminologia da ISO 13485:2016
Isto garante que os fabricantes adotem normas reconhecidas a nível mundial.
![]()
Gestão de riscos:
Enfatiza a gestão de riscos ao longo de todo o ciclo de vida do dispositivo médico.
![]()
Conceção e comandos do dispositivo
Nos termos FDA , os controlos de conceção foram alargados para garantir que os fabricantes tenham plenamente em conta as necessidades dos utilizadores, a segurança do dispositivo e os critérios de desempenho, o que constitui uma área de grande destaque durante USFDA
![]()
Vigilância Pós-Comercialização
As empresas precisam de reforçar o sistema de monitorização pós-comercialização. Isto implicará que os fabricantes recolham informações sobre a segurança e a eficácia dos dispositivos, o que ajudará a identificar e resolver rapidamente os problemas.
![]()
Documentação e manutenção de registos
A norma definitiva salienta que a documentação deve ser exaustiva, sendo fundamental manter registos rigorosos durante a inspeção.
Datas importantes
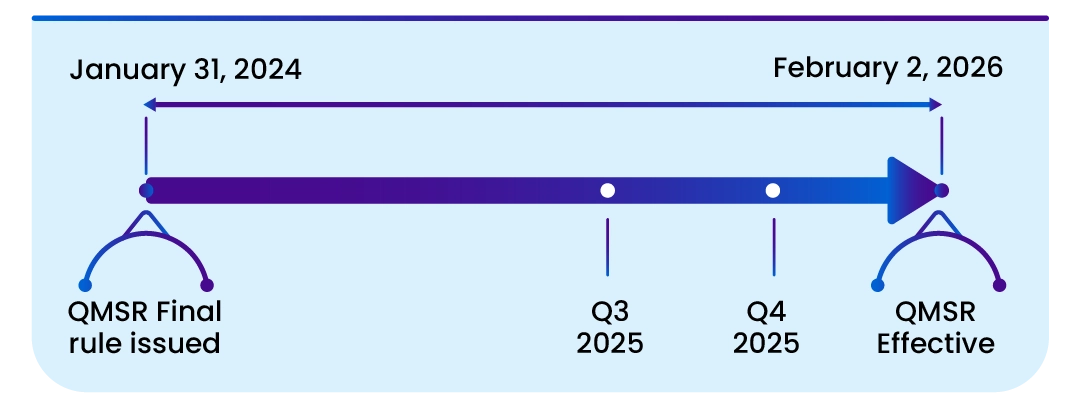
Esta norma entra em vigor a partir de 2 de fevereiro de 2026. A incorporação por referência de determinados documentos enumerados nesta norma foi aprovada pelo Diretor do Registo Federal em 2 de fevereiro de 2024.
Obtenha aconselhamento especializado sobre a sua conformidade com a QMSR
Por que razão deve seguir o quadro FDA da FDA ?
![Inteligência Regulamentar (RI)]() O QMSR entrará em vigor a partir de 2 de fevereiro de 2026, sendo obrigatório que os fabricantes atualizem o seu Sistema de Gestão da Qualidade.
O QMSR entrará em vigor a partir de 2 de fevereiro de 2026, sendo obrigatório que os fabricantes atualizem o seu Sistema de Gestão da Qualidade.![Um único parceiro para tudo]() A implementação atempada e uma manutenção eficaz podem reduzir a probabilidade de recolha de produtos e reclamações.
A implementação atempada e uma manutenção eficaz podem reduzir a probabilidade de recolha de produtos e reclamações.![garantindo a precisão das submissões]() A conformidade com a QMSR poderá reduzir as observações daFDA US ,FDA não afetará o estatuto do produto no mercado regulamentado.
A conformidade com a QMSR poderá reduzir as observações daFDA US ,FDA não afetará o estatuto do produto no mercado regulamentado.
 O QMSR entrará em vigor a partir de 2 de fevereiro de 2026, sendo obrigatório que os fabricantes atualizem o seu Sistema de Gestão da Qualidade.
O QMSR entrará em vigor a partir de 2 de fevereiro de 2026, sendo obrigatório que os fabricantes atualizem o seu Sistema de Gestão da Qualidade. A implementação atempada e uma manutenção eficaz podem reduzir a probabilidade de recolha de produtos e reclamações.
A implementação atempada e uma manutenção eficaz podem reduzir a probabilidade de recolha de produtos e reclamações. A conformidade com a QMSR poderá reduzir as observações daFDA US ,FDA não afetará o estatuto do produto no mercado regulamentado.
A conformidade com a QMSR poderá reduzir as observações daFDA US ,FDA não afetará o estatuto do produto no mercado regulamentado.Opte pelos serviços de consultoria da Freyr FDA , onde os nossos consultores de excelência o acompanharão meticulosamente em todas as fases do ciclo de vida do seu dispositivo, para garantir uma implementação perfeita dos requisitos da QMSR!
Obtenha aconselhamento especializado sobre a sua conformidade com a QMSR
QMSR
- Classificação de dispositivos médicos de acordo comFDA.
- Criação de documentos em conformidade com 21 CFR 820.
- Análise das lacunas dos documentos existentes do Sistema de Gestão da Qualidade (SGQ), em conformidade com 21 CFR 820.
- Plano de remediação para a conformidade com 21 CFR 820.
- Auditorias simuladas.
- Uma equipa dedicada de Garantia de Qualidade (QA) de especialistas em dispositivos médicos.
- Experiência comprovada na gestão da conformidade com 21 CFR 820.
- Modelos flexíveis de entrega de projetos.