4 min de leitura
A Notificação de Dispositivos Médicos (MDR) é uma ferramenta de vigilância pós-comercialização que a Food and Drug Administration (FDA) utiliza para monitorizar o desempenho dos dispositivos, detetar potenciais problemas de segurança relacionados com os dispositivos e contribuir para as avaliações de risco-benefício dos dispositivos. O objetivo do MDR é detetar e abordar eventos adversos relacionados com dispositivos de forma atempada. Permite que médicos, unidades de saúde, fabricantes e consumidores façam notificações voluntárias para compreender a segurança e eficácia do dispositivo após a sua comercialização.
O MDR é aplicável a todas as classes de dispositivos médicos, que são fabricados nos Estados Unidos da América (US) ou importados para os US. Os fabricantes de dispositivos médicos que pretendam comercializar os seus dispositivos nos US devem cumprir o MDR, caso contrário, podem incorrer em penalidades financeiras. É aplicável nos US, incluindo um evento estrangeiro, ou seja, é aplicável a dispositivos médicos legalmente comercializados nos Estados Unidos, tanto os fabricados nos US como em países estrangeiros. Além disso, existem vários casos de aplicabilidade para um MDR, tais como:
- se um dispositivo for fabricado nos US, distribuído localmente e para outros mercados
- quando um dispositivo é fabricado nos EUA, mas distribuído noutros mercados
- quando um dispositivo é fabricado no país estrangeiro, fornecido nos US e noutros mercados
- quando um dispositivo é fabricado num país estrangeiro e distribuído localmente e
- quando um dispositivo está sob investigação nos EUA
MDR e o Fluxo do Processo de Notificação
O regulamento da MDR contém muitos requisitos obrigatórios para fabricantes, importadores e instalações de utilizadores de dispositivos notificarem a FDA sobre certos eventos adversos relacionados com dispositivos e problemas de produtos. O fluxograma do processo fornecido abaixo detalha o processo de notificação passo a passo.
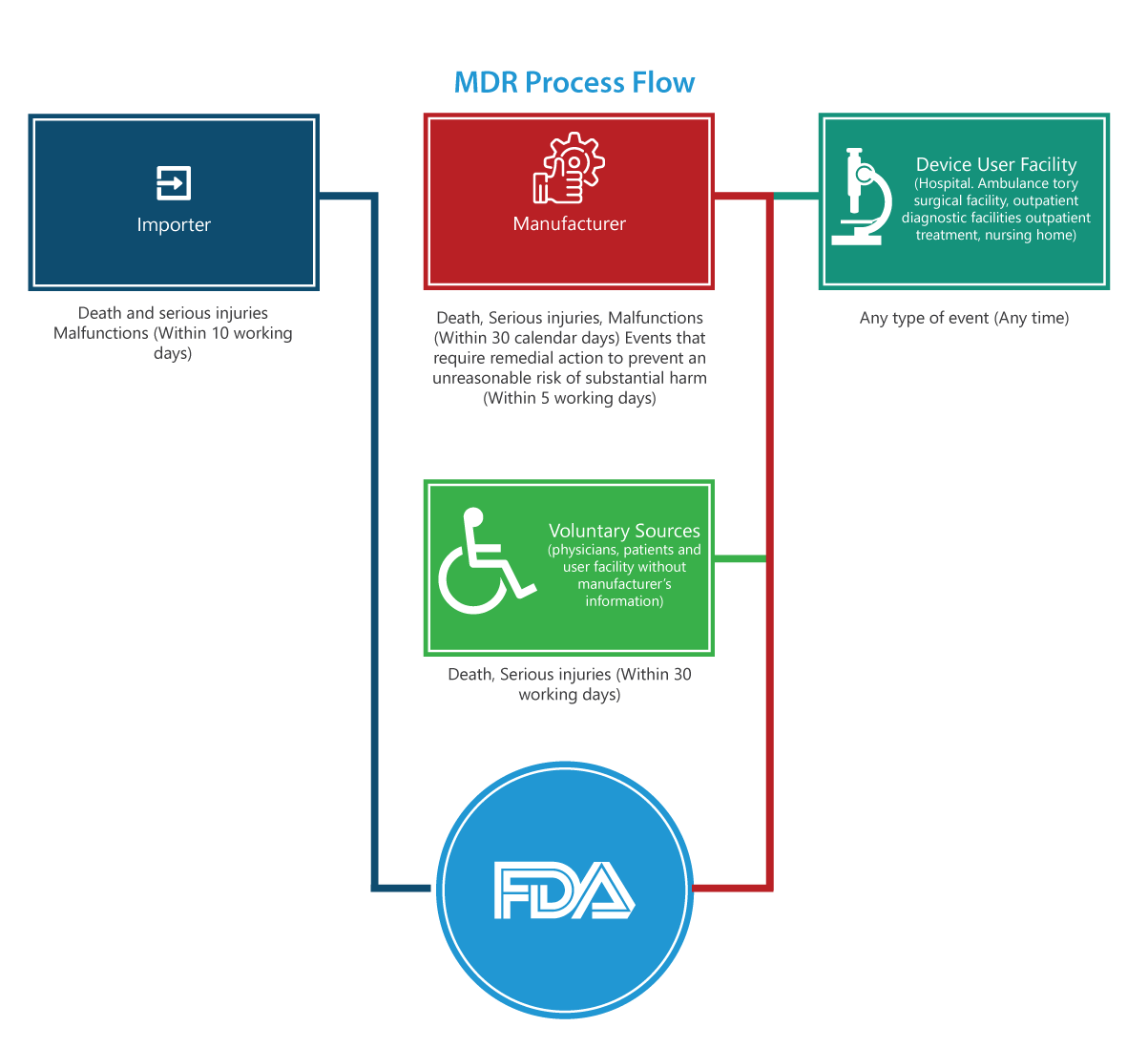
A quem se aplica?
Importadores
Os relatórios de óbitos, lesões graves e avarias devem ser submetidos à FDA e ao fabricante no prazo de 30 dias úteis. Se a avaria puder causar lesões ou óbitos noutros locais, os importadores devem notificar o fabricante sobre a avaria.
Fabricantes
Os relatórios de um evento (óbitos, lesões graves e avarias) designado pela FDA ou de um evento que exija uma ação corretiva para prevenir um risco irrazoável de dano substancial à saúde pública devem ser submetidos à FDA no prazo de 5 dias úteis, preenchendo o formulário 3500A.
Instalação de Utilizador de Dispositivo (Hospital, unidade cirúrgica ambulatorial, lar de idosos, unidade de diagnóstico ambulatorial ou unidade de tratamento ambulatorial)
Os relatórios devem ser submetidos ao fabricante do dispositivo o mais tardar 10 dias úteis após o dia em que toma conhecimento de informações de que um dispositivo causou ou pode ter contribuído para uma lesão grave num paciente da instalação. Se o fabricante for desconhecido, a instalação deve submeter o relatório à FDA.
Grupos Voluntários
Pacientes, profissionais de saúde e consumidores que encontrem um problema relacionado com um dispositivo médico podem notificar a FDA através do MedWatch
eMDR
A FDA tornou obrigatória a MDR eletrónica (eMDR) em 2015 para identificar questões críticas de qualidade e integridade dos dados associadas à notificação de lesões graves relacionadas com todas as classes de dispositivos médicos. A eMDR é o modo de notificação preferencial.
Os fabricantes podem submeter a sua eMDR através de um Gateway de Submissões Eletrónicas (ESG). A partir do momento da submissão, o gateway eletrónico demora até 48 horas a enviar uma confirmação. Se houver algum erro ao submeter o relatório, aparecerá uma mensagem para fazer as correções.
eMDR – Como é benéfico?
O eMDR oferece múltiplas vantagens em relação ao mecanismo de comunicação manual (MDR). Alguns benefícios notáveis para fabricantes, agências e pacientes estão listados abaixo:
- A ferramenta de submissão eMDR melhora a colaboração entre uma organização, a agência de saúde (FDA) e os pacientes.
- O eMDR poupa custos. A automação reduz a necessidade de despesas administrativas e de comunicação tradicional; ajuda a acelerar o processo e promove uma comunicação eficaz de eventos, resultando numa interação imediata com a FDA.
- Os processos manuais envolvem uma quantidade substancial de papelada, podem ser demorados e difíceis de monitorizar e processar. A submissão eMDR é automatizada e centralizada. Os registos podem ser recuperados facilmente, poupando muito tempo durante a revisão.
- O eMDR permite que as partes assinalem erros de submissão rapidamente, em contraste com as correspondências manuais e demoradas com a FDA.
- O eMDR atua como um único ponto de entrada para processar todas as submissões eletrónicas num ambiente altamente seguro, e é benéfico porque as reclamações na organização podem ser ligadas diretamente ao formulário MedWatch e integradas no portal da FDA.
eMDR e o Fluxo do Processo de Comunicação
O regulamento eMDR contém requisitos obrigatórios para que fabricantes, importadores e instalações de utilizadores de dispositivos comuniquem certos eventos adversos relacionados com dispositivos e problemas de produtos à FDA. O fluxograma abaixo detalha o processo de comunicação passo a passo.
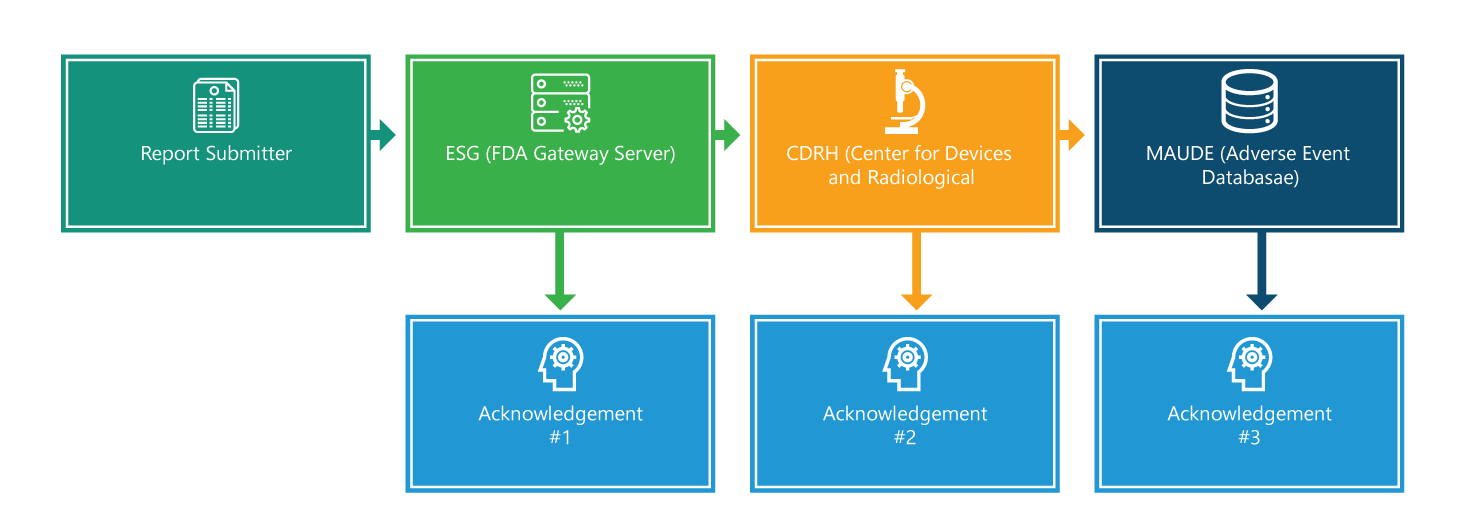
O processo de comunicação é composto por quatro etapas. Exceto a primeira, cada etapa é confirmada. Além disso, cada etapa é acompanhada de informações adicionais que ajudarão a facilitar o processo.
Etapa 1: Remetente do Relatório
Submeter um eMDR. Inicialmente, para fazer uma submissão, deve-se ter uma assinatura eletrónica e deve-se garantir que os nomes dos ficheiros de submissão incluem apenas um ponto, que é usado para indicar a extensão do tipo de ficheiro (por exemplo, 555.xml ou 555.pdf). No entanto, o tempo de entrega e processamento da submissão depende do tamanho total da sua submissão; submissões maiores demoram mais tempo a serem entregues e processadas.
Etapa 2: Portal de Submissões Eletrónicas (ESG)
Quando a sua submissão chega ao ESG, deverá receber rapidamente uma confirmação #1, a menos que o ESG esteja em manutenção. É necessário verificar o estado do seu MDR no site do ESG.
Etapa 3: CRDH
O eMDR é automaticamente encaminhado do ESG para o Center for Devices and Radiological Health (CDRH). Uma vez encaminhado, tal como na etapa 2, deverá receber uma confirmação, ou seja, #2.
Etapa 4: Experiência de Dispositivos de Fabricantes e Instalações de Utilizadores (MAUDE)
Quando o CDRH valida e atualiza a submissão na base de dados de Eventos Adversos (MAUDE), espera-se que o remetente receba uma confirmação #3. Deve-se notar que quaisquer erros que ocorram durante a validação e o carregamento são registados.
A Comunicação de Dispositivos Médicos (MDR) é um processo crítico que ajuda a salvar vidas e a proteger os pacientes de riscos desnecessários. Garante que todas as partes envolvidas nos cuidados ao paciente são responsáveis e atentas na utilização de dispositivos.
O eMDR facilita a comunicação, mas a documentação e o acompanhamento podem consumir muitos recursos. Faça-o corretamente à primeira; fale connosco em sales@freyrsolutions.com.