

4 min de leitura
Com a USFDA a implementar a regra final para os regulamentos do sistema de gestão da qualidade (QMSR) em 2024, os fabricantes de dispositivos médicos devem adotar as alterações para comercializar e distribuir os seus dispositivos no mercado dos EUA.
Esta regra atualiza os Regulamentos do Sistema de Qualidade (QSR) da USFDA, alinhando-os com a ISO 13485:2016, que é a norma internacional para SGQ de dispositivos médicos. Os fabricantes de dispositivos médicos têm um período de transição de dois anos para estarem em conformidade, o que torna necessário que as organizações adaptem os novos requisitos para evitar a não conformidade no momento da inspeção.
O que é o QMSR?
O QMSR da FDA é uma abordagem simplificada aos requisitos de QMS, que é uma atualização da antiga estrutura QSR. Este alinhamento é crucial porque simplifica a conformidade global para os fabricantes, especialmente aqueles que operam globalmente. Esta harmonização permitirá às empresas cumprir os requisitos regulamentares tanto nos US como noutros mercados de uma forma muito mais unificada.
O QMSR exige melhorias na gestão de risco, conceção de dispositivos e Vigilância Pós-Comercialização. Este papel pode adicionar mais complexidade, mas também permite aos fabricantes padronizar os seus procedimentos de qualidade, o que, por sua vez, aumenta a segurança do dispositivo e melhora a documentação, o que pode ser vital durante as inspeções da USFDA.
Principais alterações no QMSR
- Harmonização com a ISO 13485: Este é o passo mais crítico que permite aos fabricantes de dispositivos médicos aceitar padrões internacionalmente reconhecidos. A USFDA reconheceu que muitas empresas de dispositivos médicos já estão em conformidade com a ISO 13485, o que reduz esforços duplicados.
- A gestão de risco destaca a importância de gerir os riscos ao longo de todo o ciclo de vida do dispositivo médico. Os fabricantes de dispositivos médicos devem demonstrar uma gestão e controlo de risco eficazes.
- Conceção e controlos de dispositivos: Ao abrigo do QMSR, os controlos de conceção foram alargados para garantir que os fabricantes de dispositivos médicos consideram plenamente as necessidades do utilizador, a segurança do dispositivo e os critérios de desempenho, o que é uma área de foco durante a inspeção da USFDA.
- Vigilância Pós-Comercialização: As empresas precisam de melhorar o sistema de monitorização pós-comercialização. Isto implicará que os fabricantes recolham informações sobre a segurança e eficácia do dispositivo, o que ajuda a identificar e resolver os problemas rapidamente.
- Documentação e manutenção de registos: Esta é a regra final que enfatiza a documentação. A manutenção de registos completa/adequada é fundamental durante a inspeção.
Etapas para preparar a inspeção da US Food and Drug Administration:
Com a inspeção a decorrer até 2026, as empresas têm dois anos para adaptar os seus sistemas de qualidade ao QMSR. No entanto, esperar pelo último minuto pode ser arriscado.
Passos para a inspeção da USFDA sob o quadro QMSR para indústrias onde o SGC atual se baseia no QSR:
- Realizar uma análise de lacunas: Este é o primeiro passo onde o sistema de qualidade atual diverge dos novos requisitos do QMSR. Uma análise de lacunas completa ajudará a identificar as áreas que necessitam de atualizações, como gestão de riscos, vigilância pós-comercialização e controlos de conceção.
- Atualizar o procedimento de gestão de risco: Garantir que as atividades de gestão de risco estejam integradas em todo o ciclo de vida do seu produto, desde a conceção até à Vigilância Pós-Comercialização.
- Rever o controlo de conceção: Os fabricantes devem garantir que o processo de conceção é robusto e bem documentado. Validar que o processo de conceção é robusto, bem documentado e totalmente integrado no seu sistema de gestão de qualidade.
- Melhore a Vigilância Pós-Comercialização: Implemente sistemas para monitorizar o desempenho do dispositivo após a sua colocação no mercado. Isso pode envolver a criação de mecanismos de feedback do cliente, a recolha de evidências de dados clínicos e o seu acompanhamento rigoroso.
- Formação e documentação: Formar o pessoal sobre os novos requisitos, especialmente aqueles que estão envolvidos na gestão da qualidade e conformidade regulamentar. Isto garante que todo o processo de documentação está alinhado com as expectativas do QMSR.
- Certificação por terceiros: Se a sua empresa não possui a certificação ISO 13485, agora pode ser o momento de a considerar. Obter a certificação ISO 13485 pode dar-lhe uma vantagem no cumprimento dos requisitos da USFDA para aumentar a credibilidade nos mercados globais.
A navegação pelo período de transição de dois anos
Para utilizar eficazmente o período de transição e garantir a conformidade com o Regulamento do Sistema de Gestão da Qualidade (QMSR) da USFDA, os fabricantes devem adotar uma abordagem proativa. Aqui está um roteiro sugerido:
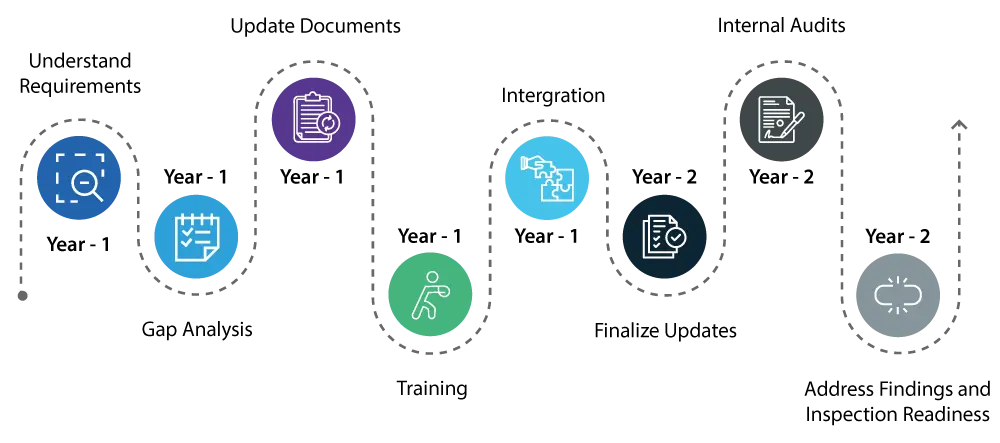
Ano 1:
- Compreender o requisito: Comece por compreender detalhadamente as alterações introduzidas pelo QMSR. Isto implica uma revisão detalhada dos novos requisitos e de como estes diferem do Regulamento do Sistema de Qualidade (SQ) existente.
- Identifique as lacunas : Realize uma análise de lacunas abrangente para identificar áreas no seu sistema de qualidade atual que necessitam de atualizações para cumprir os novos padrões QMSR.
- Atualização / Remediação de Documentos: Iniciar as atualizações necessárias ao seu sistema de qualidade, focando-se em áreas como a gestão de riscos e os controlos de design, que são componentes críticos do QMSR.
- Formação: Comece a formar o seu pessoal sobre os novos regulamentos para garantir que todos os envolvidos estão cientes das alterações e compreendem os seus papéis na manutenção da conformidade.
- Integração: Comece a integrar os novos requisitos QMSR nas suas operações diárias para tornar a transição mais suave.
Ano 2:
- Prossiga com a implementação de alterações no seu sistema de qualidade, garantindo que todas as atualizações estejam totalmente integradas e operacionais.
- Realizar auditorias internas exaustivas para verificar se as atualizações são eficazes e se o seu sistema de qualidade está totalmente alinhado com os requisitos do QMSR.
- Aborde prontamente quaisquer resultados das auditorias internas para garantir que todos os aspetos do seu sistema de qualidade estão em conformidade.
- Até ao final do segundo ano, o seu sistema de qualidade deverá estar totalmente em conformidade com o QMSR, e deverá estar preparado para as inspeções da USFDA com a confiança de que não haverá grandes problemas.
Ao seguir este roteiro, os fabricantes podem não só cumprir os requisitos da USFDA, mas também estabelecer um sistema de qualidade robusto que seja eficiente, padronizado e reconhecido globalmente. Esta abordagem proativa ajudará a garantir uma transição suave para os novos regulamentos e a manter os mais altos padrões de qualidade e segurança para dispositivos médicos.
Conclusão: Adotar Medidas Proativas
Preparar-se para as inspeções da USFDA ao abrigo da nova regra QMSR não se trata apenas de evitar penalidades — trata-se de melhorar a segurança e a eficácia do dispositivo. Ao harmonizar com a ISO 13485, a USFDA está a definir expectativas mais elevadas, mas também a fornecer um caminho para uma conformidade global mais simplificada. Os fabricantes que começarem a adaptar-se cedo devem focar-se em áreas-chave como a gestão de riscos e a vigilância pós-comercialização, e garantir que os seus sistemas de qualidade estão atualizados e robustos, o que não só irá satisfazer as expectativas regulamentares, mas também melhorar a sua vantagem competitiva no mercado.
