2 min de leitura
Shonin (Aprovação Pré-Mercado) é o percurso regulamentar para o registo de dispositivos médicos no Japão. O percurso Shonin destina-se principalmente ao registo de dispositivos médicos de Classe II e III para os quais as normas de classificação da PMDA não estão disponíveis. Para os dispositivos de Classe IV de alto risco, os fabricantes também devem apresentar a submissão Shonin. A PMDA é responsável pela revisão e aprovação da submissão Shonin.
Quais São as Outras Vias de Registo de Dispositivos no Japão?
Além de Shonin, as vias Todokede e Ninsho são também utilizadas para aprovações de dispositivos médicos no Japão. Os fabricantes de dispositivos médicos podem escolher uma delas, dependendo da classe de risco do dispositivo e da disponibilidade de precedentes no Japão. O fabricante deve identificar a classificação do dispositivo e pesquisar a disponibilidade da Norma Industrial Japonesa (JIS) antes de determinar a via de registo aplicável.
- Todokede (Submissão Pré-Mercado) – É aplicável a dispositivos de Classe I e exige que os fabricantes submetam uma notificação pré-mercado à PMDA para aprovação.
- Ninsho (Certificação Pré-Mercado) - É aplicável a dispositivos genéricos de Classe II e III que possuem padrões de certificação (Normas JIS). O Organismo de Certificação registado (RCB) é responsável pela revisão e aprovação da submissão.
Quais São os Pré-requisitos para o Registo Shonin?
Os fabricantes que registam os seus dispositivos através da via Shonin devem planear meticulosamente as submissões. Devem garantir o seguinte:
- A submissão de dados gerais do dispositivo, tais como a categoria do dispositivo médico, a utilização prevista, dados de análise de risco de eficácia, dados clínicos, entre outros.
- Fornecimento do Resumo da Documentação Técnica (STED)
- Fornecimento de documentos apenas em língua japonesa
- Os fabricantes estrangeiros devem obrigatoriamente nomear um Titular da Autorização de Introdução no Mercado (MAH) ou um Titular da Autorização de Introdução no Mercado Designado (DMAH)
- Os fabricantes estrangeiros devem obter o certificado de Registo de Fabricante Estrangeiro (FMR) para os seus estabelecimentos de fabrico.
Quais são os requisitos de QMS para o Registo de Dispositivos no âmbito do processo Shonin?
Os fabricantes devem cumprir todos os requisitos do SGQ definidos na Portaria 169. O patrocinador, o DMAH ou o MAH devem apresentar um pedido à PMDA. A PMDA realiza uma inspeção detalhada do SGQ nas instalações do fabricante e emite o certificado após a implementação satisfatória do SGQ.
Qual é o processo de Registo para a aprovação de dispositivos no âmbito do processo Shonin?
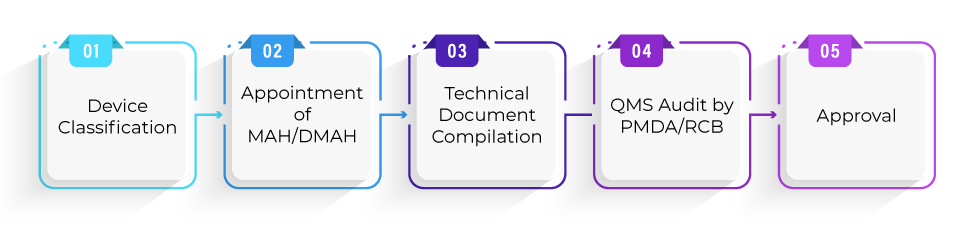
Qual é o prazo médio necessário para a aprovação de dispositivos no âmbito do processo Shonin?
A PMDA geralmente requer 12 meses para a avaliação técnica a partir da data de receção da submissão Shonin. O fabricante deve considerar o tempo necessário para preparar os documentos de submissão ou realizar estudos clínicos nos seus cronogramas de projeto.
Existe algum Prazo de Validade para o registo de dispositivos ao abrigo da via Shonin?
O registo de dispositivos médicos não expira, mas o patrocinador deve renovar os certificados de QMS a cada cinco (05) anos.
O Japão é um mercado lucrativo mas que, intrinsecamente, apresenta complexidades regulamentares e barreiras linguísticas. Os fabricantes devem considerar estes fatores e planear proativamente a sua estratégia de entrada no mercado (GTM) para o Japão. Os fabricantes de dispositivos médicos e IVD podem optar por terceirizar todas as nuances regulamentares para um parceiro regulamentar fiável e utilizar os recursos para se concentrarem noutros componentes essenciais.
Para saber mais sobre a aprovação de dispositivos médicos Shonin no Japão ou quaisquer outras regulamentações da PMDA Japão, contacte hoje os especialistas em regulamentação da Freyr.