
3 min de leitura
No setor da produção de dispositivos médicos, em constante evolução e altamente regulamentado, garantir a qualidade dos produtos e a segurança dos doentes não é apenas uma obrigação legal, mas também um imperativo estratégico. Sistemas eficazes de gestão de reclamações e de vigilância, em conformidade com ISO 13485:2016, constituem a espinha dorsal de qualquer Sistema de Gestão da Qualidade (SGQ) robusto. Estes mecanismos asseguram a conformidade com as regulamentações globais e reforçam a confiança dos clientes e a credibilidade da marca.
Vamos analisar como o tratamento de reclamações e a vigilância desempenham um papel fundamental na manutenção da qualidade e da conformidade regulamentar no setor dos dispositivos médicos, com informações alinhadas com ISO 13485:2016 e apoiadas por quadros regulamentares globais.
Compreender a gestão de reclamações ao abrigo da ISO 13485:2016
ISO 13485:2016 define uma reclamação como qualquer comunicação escrita, oral ou eletrónica que alegue falhas relacionadas com a identidade, qualidade, durabilidade, fiabilidade, usabilidade, segurança ou desempenho de um dispositivo médico. Esta norma exige que as empresas de dispositivos médicos estabeleçam procedimentos documentados para o tratamento de reclamações, incluindo a avaliação, investigação e resolução das mesmas, como parte de um sistema eficaz de gestão da qualidade.
Um sistema de gestão de reclamações é fundamental para garantir a conformidade, manter a satisfação do cliente e melhorar a qualidade dos dispositivos. O processo deve ser organizado e transparente, envolvendo as seguintes etapas:
- Registo e documentação de reclamações
Todas as reclamações devem ser registadas sem demora, com o máximo de detalhes possível sobre a natureza do problema, as especificações e o modelo do dispositivo, as condições de utilização e quaisquer consequências sentidas pelo utilizador ou paciente. - Avaliação da obrigatoriedade de notificação
Após o seu registo, as reclamações devem ser avaliadas para determinar se constituem incidentes sujeitos a notificação. Por exemplo, nos EUA, os fabricantes de dispositivos médicos são obrigados a notificar determinados problemas com os dispositivos ao abrigo do sistema de Notificação de Dispositivos Médicos (MDR), regulamentado pela FDA. Os fabricantes devem cumprir os prazos de notificação previstos na regulamentação aplicável. - Investigação e Análise da Causa Raiz
As reclamações válidas devem ser seguidas de uma investigação detalhada. Identificar a causa raiz é essencial para implementar ações corretivas e preventivas (CAPA) eficazes. - Ações corretivas e preventivas (CAPA)
Com base nas conclusões relativas aos dispositivos médicos, as empresas devem implementar CAPAs para resolver tanto o problema imediato como evitar a sua recorrência e prevenir ocorrências futuras. - Ciclo de feedback e encerramento
Assim que o problema for resolvido, a reclamação deve ser formalmente encerrada, com documentação completa e comunicação ao reclamante, se aplicável ao dispositivo médico.
Vigilância: um pilar fundamental da vigilância pós-comercialização
A vigilância refere-se ao acompanhamento e à comunicação de eventos e incidentes adversos após a colocação de um dispositivo médico no mercado. De acordo com ISO 13485:2016 e os regulamentos globais correspondentes, esta vigilância contínua é essencial para a deteção precoce de riscos potenciais e a implementação de intervenções atempadas.
O sistema de vigilância da União Europeia, tal como atualizado ao abrigo do Regulamento relativo aos dispositivos médicos (EU MDR), exige que os fabricantes de dispositivos médicos disponham de planos de vigilância pós-comercialização. Estes planos devem incluir processos para a recolha e análise de dados relativos à utilização dos dispositivos na prática.
Da mesma forma, na Índia, a Central Drugs Standard Control Organization CDSCO) regula os processos de vigilância e de reclamação relativos aos dispositivos médicos. Esta entidade incentiva a comunicação de eventos adversos e de falhas nos produtos, com o objetivo de melhorar continuamente a segurança e a eficácia dos dispositivos médicos.
Os elementos-chave de um sistema de vigilância eficaz incluem:
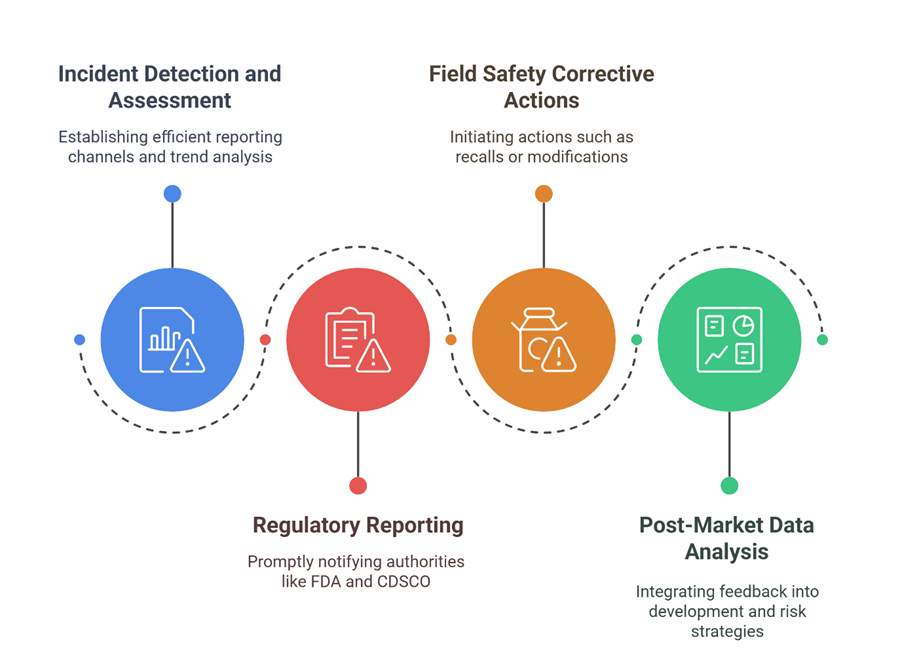
Desenvolvimento de uma estratégia integrada de reclamações e vigilância
Os fabricantes de dispositivos médicos podem melhorar o seu sistema de gestão da qualidade (SGQ) integrando o tratamento de reclamações e a vigilância num único sistema coeso. Aqui estão algumas boas práticas:
Procedimentos operacionais padrão (SOP) e definições de funções claras
Os Procedimentos Operacionais Padrão (POP) devem definir as etapas para a receção, avaliação, comunicação e resolução de reclamações. Definem quem é o responsável em cada fase, a fim de evitar atrasos e lacunas.
- Formação e sensibilização dos funcionários
A formação regular dos funcionários garante que o pessoal compreenda como identificar e lidar com reclamações e eventos adversos de forma eficaz. - Ferramentas digitais e automatização
A utilização de software de gestão da qualidade (QMS) permite simplificar o acompanhamento de reclamações, a documentação e a elaboração de relatórios. A automatização melhora a rastreabilidade e a conformidade com ISO 13485:2016. - Cultura da Qualidade e Melhoria Contínua
Promover uma filosofia que valorize o feedback incentiva a comunicação proativa de problemas e estimula a inovação nas melhorias de qualidade.
Conformidade e muito mais: uma vantagem de marketing
A implementação de sistemas em conformidade com ISO 13485:2016 para a gestão de reclamações e vigilância não só garante o cumprimento da regulamentação, como também posiciona a empresa como uma entidade de confiança e focada na qualidade. Num setor em que a segurança e o desempenho têm um impacto direto nas vidas das pessoas, esse compromisso pode constituir uma poderosa ferramenta de marketing.
De acordo com FDA a conformidade com as normas internacionais melhora o acesso aos mercados globais, reforça a confiança dos clientes e reduz o risco de recalls dispendiosos ou de litígios.
| Região | Quadro Regulatório | N.º da cláusula de referência/artigo para o tratamento de reclamações | N.º da cláusula de referência / Artigo relativo à vigilância / Notificação de eventos adversos |
| União Europeia | EU MDR 2017/745 | Artigo 83.º, Anexo III (sistema PMS); n.º 9 do artigo 10.º | Artigos 87.º a 92.º (Sistema de Vigilância), Anexo III |
| Estados Unidos (FDA) | 21 CFR Parte 820 (QSR) | §820.198 – Processos de reclamação | 21 CFR Parte 803 – Notificação de Dispositivos Médicos (MDR) |
| Índia (CDSCO) | Regulamento dos Dispositivos Médicos de 2017 (MDR) | Capítulo VII, Regras 25 e 26 | Capítulo VII, Regra 27 |
| Global (Norma ISO) | ISO 13485:2016 | Cláusula 8.2.2 – Tratamento de reclamações | Cláusula 8.2.3 – Comunicação às autoridades reguladoras |
Conclusão
No atual panorama regulamentar competitivo dos dispositivos médicos, a gestão eficaz das reclamações e os sistemas de vigilância são imprescindíveis. Em conformidade com ISO 13485:2016, estes processos não só protegem os doentes como também contribuem para a construção de uma marca resiliente e orientada para a reputação. Ao investir em procedimentos sistemáticos, formação contínua e vigilância pós-comercialização, os fabricantes podem transformar os requisitos regulamentares em oportunidades de crescimento e liderança de mercado.









