

3 min de leitura
No mundo em evolução dos dispositivos médicos, a conformidade não é uma tarefa única — é um compromisso contínuo. A monitorização contínua e as atualizações de relatórios chave, como os Relatórios de Avaliação Clínica (CERs), Relatórios de Avaliação de Desempenho (PERs) e Relatórios Periódicos de Atualização de Segurança (PSURs), são cruciais ao longo de todo o ciclo de vida de um dispositivo médico, desde a investigação inicial até à vigilância pós-comercialização. À medida que o panorama dos avanços médicos e dos requisitos regulamentares evolui continuamente, garantir a segurança e a conformidade dos dispositivos médicos e IVDs através de uma redação médica eficaz continua a ser a pedra angular do sucesso e da viabilidade a longo prazo.
Vejamos como a gestão do ciclo de vida continua a ser essencial para o sucesso de um dispositivo médico.
O lançamento de um dispositivo médico é o culminar de anos de esforço dedicados a várias fases, como investigação, desenvolvimento, ensaios clínicos, submissões regulamentares e vigilância pós-comercialização. Este processo abrange muitos anos e cada fase acumula dados vitais em grande quantidade, que devem ser cuidadosamente compilados e analisados para garantir que o dispositivo permanece seguro.
A conformidade não é um evento único, e uma gestão eficaz do ciclo de vida ajuda a evitar atrasos que podem ser dispendiosos mais tarde. Estes atrasos ocorrem frequentemente devido a um planeamento ineficaz para alterações regulamentares imprevistas ou não conformidade, que podem ter sido negligenciadas. Atualizações regulares de documentos críticos como CERs, PERs e PSURs garantem que os fabricantes cumprem os requisitos regulamentares em evolução e mantêm a segurança do produto. A gestão do ciclo de vida garante que os fabricantes, desde o início, planearam cada etapa, e assim podem evitar quaisquer armadilhas e assegurar uma entrada bem-sucedida no mercado e longevidade para os seus dispositivos.
Está a par da conformidade?
Desde ligaduras a implantes, a indústria de dispositivos médicos é um setor de ponta, criativo e com uma riqueza de perspetivas. Apesar do forte desejo dos fabricantes de fornecer ao mercado produtos seguros e de alta qualidade, a ambiguidade persiste. O EU MDR substituiu a Diretiva de Dispositivos Médicos (MDD) e a Diretiva de Dispositivos Médicos Ativos Implantáveis (AIMDD). Este regulamento foi implementado para impor controlos mais rigorosos, melhorar a segurança do paciente e promover uma maior transparência no setor da saúde. Existe muita incerteza sobre essas normas, e questões cruciais de conformidade são frequentemente negligenciadas. No entanto, os fabricantes de dispositivos médicos devem tomar medidas para evitar dificuldades regulamentares.
Vamos investigar os desafios mais comuns enfrentados pelos fabricantes de dispositivos médicos.
- Manutenção de CAPA (Corrective and Preventive Action)
- Aderir aos Procedimentos de Reclamação
- Seguimento dos Procedimentos de Vigilância
- Avaliação Clínica e Vigilância Pós-Comercialização
- Ligação com organismos notificados
Boas práticas para manter a conformidade
Para superar estes desafios, nós, na Freyr, estabelecemos
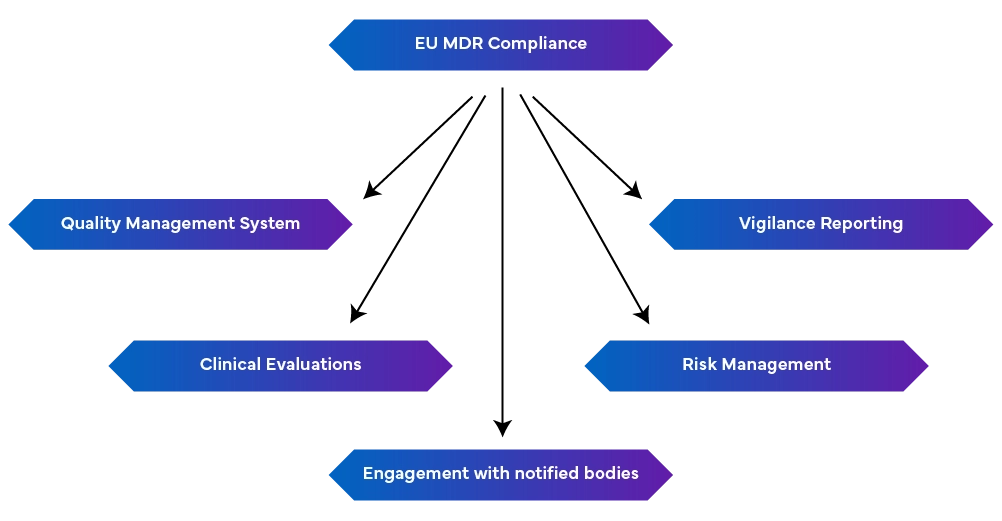
-um Sistema de Gestão da Qualidade Robusto (EN ISO 13485:2016): Isto inclui aspetos de produção, incluindo conformidade regulamentar, documentação técnica, declarações de conformidade da UE e gestão de riscos.
-Notificação de Vigilância: Manter a notificação de vigilância como um processo contínuo, em vez de um esforço pontual.
- Gestão Proativa de Riscos: Isto identifica e mitiga riscos ao longo do ciclo de vida do dispositivo, atualizando regularmente para abordar riscos emergentes.
- Realizar Avaliações Clínicas e Vigilância Pós-Comercialização: Isto é feito demonstrando a segurança e o desempenho do dispositivo usando dados clínicos. Se necessário, devem ser realizadas investigações clínicas, e as avaliações clínicas devem ser regularmente atualizadas com dados de vigilância pós-comercialização. Relatórios periódicos de atualização de segurança que resumem as descobertas também são necessários.
- Ligação com Organismos Notificados: As alterações nos regulamentos podem afetar os requisitos do MDR, tornando crucial manter um bom relacionamento com os Organismos Notificados para atualizações e modificações rápidas.
Como um especialista em regulamentação o pode ajudar
Manter-se atualizado com um panorama regulamentar em constante mudança pode ser difícil. Então, por que não deixar que um especialista o guie através deste labirinto? Gerir as exigências regulamentares do ciclo de vida de um dispositivo médico não é apenas complexo, mas também intensivo em recursos. No caso de empresas mais pequenas, onde os recursos internos podem ser melhor utilizados noutras áreas, a ajuda de um parceiro regulamentar externo pode revelar-se indispensável. Eles oferecem conhecimento especializado sobre regulamentos em evolução e garantem que os seus relatórios chave — como CERs, PERs e PSURs — são continuamente atualizados e estão em conformidade.
Pode evitar atrasos dispendiosos, prevenir a não conformidade e otimizar o seu processo de aprovação. Além disso, um especialista em regulamentação pode oferecer soluções personalizadas com base nas necessidades específicas do seu dispositivo, garantindo que a vigilância pós-comercialização, a análise de lacunas e toda a documentação de conformidade estejam atualizadas. O seu envolvimento pode tornar o processo geral mais simples, minimizar os riscos e garantir que o dispositivo possa satisfazer as expectativas de segurança e desempenho.
A Freyr pode ajudá-lo com todas as suas necessidades regulamentares relativas à gestão do ciclo de vida. Contacte-nos hoje!









