3 min de leitura
O Regulamento de Dispositivos Médicos da European Union (EU MDR) 2017/745 define o âmbito dos dispositivos médicos estéreis como dispositivos que devem estar livres de microrganismos (bactérias, vírus, etc.). Os dispositivos de Classe I com função estéril também podem obter uma autocertificação, mas difere em termos de requisitos em comparação com outras subcategorias de dispositivos de Classe I.
Os requisitos para dispositivos de Classe I variam muito desde o fabrico até à colocação do dispositivo no mercado. O mesmo se aplica a dispositivos médicos de Classe I com condições estéreis. Por exemplo, se for um dispositivo de Classe I com uma função estéril, os requisitos para o fabrico e a criação de um Sistema de Gestão da Qualidade (SGQ) difeririam muito em comparação com os dispositivos de Classe I com funções de medição.
Este artigo visa fornecer uma lista de verificação concisa a considerar ao colocar um dispositivo de Classe I com condições estéreis na União Europeia. A lista de verificação apoiará os fabricantes na tomada de decisões informadas sobre diferentes aspetos.
O que fazer!
Conheça o Âmbito Antes do Fabrico
De acordo com o EU MDR, os dispositivos médicos com funções estéreis devem ser desenvolvidos e fabricados com procedimentos adequados para garantir a esterilidade até serem colocados no mercado. Os dispositivos só podem ser reutilizados após procedimentos adequados de limpeza, desinfeção e esterilização.
Escolher as Práticas de Esterilização Corretas
Existem diferentes métodos de esterilização que um fabricante pode escolher para esterilizar o dispositivo. O método de Óxido de Etileno (EtO) é amplamente utilizado para esterilizar dispositivos médicos. A escolha da esterilização deve ser devidamente justificada. Algumas normas que os fabricantes podem seguir para estabelecer, garantir e manter a esterilização são EN ISO 14937, ISO TS 21387, ISO TS 13004, ISO 11137, entre outras.
Note-se também que a documentação do método de esterilização detalhado, do método em processo e da validação de dados deve ser feita com precisão. As informações de esterilização, como temperatura de aquecimento, condições da sala, validações de processo, etc., devem ser fornecidas de acordo com os requisitos do EU MDR.
Conheça os Requisitos e as Orientações a Seguir
Um dos passos críticos é documentar os requisitos técnicos e conhecer as normas aceites ao abrigo do EU MDR. Por exemplo, a EN 556 é a norma europeia para dispositivos esterilizados. A EN 556 detalha os requisitos para designar um dispositivo médico como estéril. Além disso, a ISO TS 19930 especifica estratégias para garantir a esterilidade dos dispositivos. No entanto, ainda não foi adotada como norma da UE.
Conheça o Seu Percurso
Para obter a marca CE, esteja informado sobre o envolvimento dos Organismos Notificados e a via de avaliação da conformidade que deve ser seguida. Além disso, os dispositivos médicos em condições estéreis devem seguir os Anexos IX e XI.
O envolvimento de um Organismo Notificado é necessário, mas limitado. No caso de dispositivos estéreis, o envolvimento dos Organismos Notificados será no que diz respeito à manutenção e garantia das condições de esterilidade. Por exemplo, as avaliações de conformidade incluirão avaliações microbiológicas, métodos asséticos, etc.
O que não fazer!
Não Perca os Requisitos de Embalagem e Rotulagem

Existem requisitos adicionais para a conformidade da embalagem e rotulagem em dispositivos estéreis. Ao cumprir os requisitos de embalagem e rotulagem, não se deve esquecer a informação que deve ser afixada. O primeiro e mais importante passo é afixar um símbolo de esterilidade (mencionado abaixo) na rotulagem do dispositivo.
Deve ser incluída informação como a declaração de condição estéril, o método de esterilização, instruções em caso de embalagem estéril danificada ou aberta involuntariamente, entre outros. Além disso, a vida útil do produto estéril e o processo de reesterilização (se aplicável) devem ser incluídos. O número de identificação de um Organismo Notificado também deve ser afixado no dispositivo.
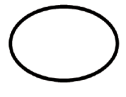
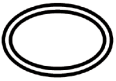
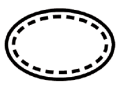
Alguns dos símbolos que devem ser adicionados (dependendo da aplicabilidade) na rotulagem de dispositivos médicos estéreis são:
Símbolo | Indicação |
| Dispositivos médicos estéreis com método de esterilização utilizado como técnica assética |
| Dispositivos médicos estéreis com método de esterilização por óxido de etileno |
| Dispositivos médicos estéreis com método de esterilização por calor seco ou vapor |
| Dispositivos médicos estéreis com método de esterilização por peróxido de hidrogénio vaporizado |
| Dispositivos médicos estéreis com método de esterilização por radiação |
| O dispositivo não pode ser esterilizado novamente. |
| O dispositivo não está esterilizado. |
| Não utilize se a embalagem estiver danificada |
| Sistema de barreira estéril único |
| Sistema de barreira estéril dupla |
| Sistema de barreira estéril único com embalagem protetora no interior |
| Sistema de barreira estéril único com embalagem protetora no exterior |


Não se esqueça dos requisitos para a colocação pós-comercialização
Os dispositivos estéreis são obrigados a manter um registo de eventos adversos em caso de qualquer ocorrência. Neste caso, os incidentes registados, como documentos, são então exigidos pelas autoridades competentes mediante solicitação.
O EU MDR tem requisitos específicos no que diz respeito à autocertificação. É um procedimento complexo, sendo o fator principal as diferentes classes dentro dos dispositivos de Classe I e os seus diferentes aspetos de requisitos para a obtenção da marcação CE. Os fabricantes de dispositivos de Classe I em condições estéreis devem estar cientes da designação do âmbito e dos requisitos específicos.
Deseja compreender em detalhe os vários aspetos dos dispositivos estéreis de classe I? Gostaria de discutir isto com um especialista comprovado? Contacte-nos.









