
3 min de leitura
Um protocolo de validação é definido como um plano documentado para testar um dispositivo médico, a fim de confirmar que o processo de produção utilizado para fabricar o produto cumpre os requisitos específicos do utilizador, técnicos e regulamentares. Isto inclui uma revisão das variáveis do processo e das limitações operacionais, bem como a análise dos resultados dos testes em condições de utilização reais.
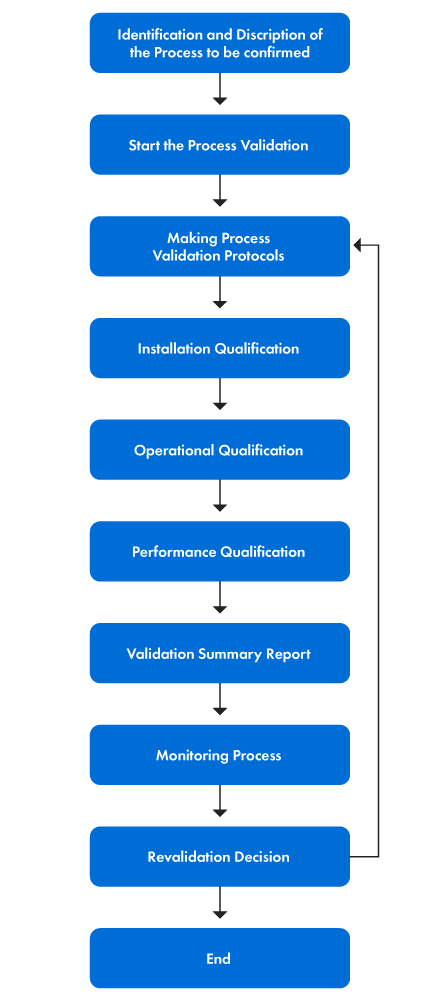
O processo de validação envolve várias ações tangíveis. Os passos são os seguintes:
- Primeiro, a equipa de validação é formada, e a cada membro são atribuídos papéis e responsabilidades específicos. O objetivo da validação do processo é fornecer uma declaração clara dos objetivos de validação e definir o âmbito das atividades de validação, especificando os aspetos do dispositivo médico que estão a ser validados. A equipa compreende então os princípios subjacentes do processo para identificar parâmetros específicos e resultados desejados.
- Segundo, os critérios de avaliação e aceitação são estabelecidos, juntamente com a seleção de métodos de teste, ferramentas e técnicas de análise estatística apropriados. Em seguida, os protocolos de validação do processo são elaborados, e a Qualificação de Instalação (QI), Qualificação Operacional (QO) e Qualificação de Desempenho (QD) são implementadas.
- Por fim, são determinados controlos de processo contínuos e medidas de monitorização para garantir a validação contínua do processo. Sempre que necessário, a revalidação é realizada para manter a precisão e a eficácia do processo de validação.
A Figura 1 abaixo apresenta uma representação passo a passo do processo de validação.
Figura 1: As Fases do Processo de Validação
PVP
Devido à vasta gama de volumes de produção e complexidades de fabrico, existem inúmeras abordagens para a realização da validação de processos. No entanto, os regulamentos da United States Food and Drug Administration (USFDA) e a ISO 13485 fornecem sugestões limitadas sobre métodos específicos. No entanto, uma fonte amplamente reconhecida e autoritária para a validação de processos de dispositivos médicos é um documento de orientação da Global Harmonization Task Force (GHTF), agora denominada International Medical Device Regulators Forum (IMDRF), publicado em 2004. Permanece a principal referência, mesmo no website oficial da USFDA.
De acordo com o documento de orientação, é formada uma equipa de validação para criar um Plano de Validação de Processos (PVP) detalhado. Os protocolos de validação de processos incluem um esquema detalhado sobre como implementar a IQ, OQ, PQ e a revalidação. O PVP deve conter os seguintes elementos:
- Definição do dispositivo e determinação da abordagem de validação.
- Identificação dos elementos que requerem validação.
- Realização de atividades no local designado.
- Delinear o âmbito da documentação.
- Criação de um cronograma para as atividades de validação.
- Desenvolvimento de um cronograma mestre geral.
- Manutenção de uma lista abrangente e referências a validações internas e externas que foram realizadas.
O protocolo de validação é redigido antes da realização das atividades de validação. Deve ser preparado pela equipa de validação e aprovado pelo departamento responsável. O objetivo de um protocolo de validação é definir os guiões de teste que devem ser seguidos para garantir que os processos e equipamentos estão prontos para fabricar produtos de dispositivos médicos seguros e eficazes.
Um relatório analítico que contém informações, juntamente com a análise, explicações e recomendações necessárias, faz parte do protocolo de validação. Estes registos são posteriormente revistos para garantir que os dois (02) critérios seguintes são cumpridos:
- Cumprimento das normas regulamentares.
- Todos os registos e dados gerados são revistos quanto aos resultados, adequação e completude.
A Figura 2 abaixo representa o PVP e os vários processos envolvidos.
Figura 2: O PVP e os seus Requisitos
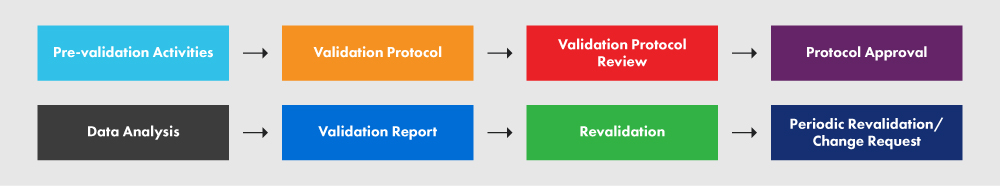
Um protocolo devidamente elaborado fornece diretrizes, políticas e procedimentos claros a serem seguidos durante a validação do processo. Abrange aspetos como instalações, equipamentos, métodos e formação. O protocolo especifica as entradas e limites do processo, bem como os passos essenciais para a execução bem-sucedida do projeto de validação do processo. Embora o seguinte esquema não abranja todos os elementos necessários no seu protocolo, oferece uma visão geral do nível de detalhe exigido. Recomendamos vivamente que siga o documento de orientação para uma melhor compreensão do processo.
- Página de Título
- Produtos a Abranger
- Equipamento/Processo a Validar
- Geral
- Objetivos
- Documentos de Referência
- Plano de Validação
- IQ
- OQ
- PQ
- Equipamento de Medição/Teste e Calibração
- Manutenção de Equipamento
- Revalidação
- Página de Aprovação/Assinatura da Equipa de Validação
A gestão de operações desempenha um papel crucial na manutenção do desempenho ideal, monitorizando as principais métricas, revendo os métodos e procedimentos de trabalho e tomando medidas rápidas quando surgem problemas. Nos casos em que existam problemas, pode ser necessário revalidar um processo parcial ou totalmente. De acordo com a Secção 820.75(c) do Regulamento do Sistema de Qualidade (QSR) da USFDA, a revalidação do processo deve ser considerada nestas circunstâncias: “Quando ocorrem alterações ou desvios no processo, o fabricante deve rever, avaliar e realizar a revalidação conforme apropriado. Estas atividades devem ser documentadas.”
Possíveis fatores para a revalidação de um processo incluem modificações às especificações, métodos, procedimentos, software, designs, componentes chave, dimensionamento de lotes, alterações de localização, alterações de equipamento e similares. Além disso, a implementação de Ações Corretivas e Preventivas (CAPA) também pode servir como um fator para a revalidação do processo. As principais razões para a revalidação são as seguintes:
- Alterações efetuadas ao processo.
- Tendência negativa na qualidade, deterioração súbita da qualidade ou um aumento acentuado nas reclamações de clientes.
- Expansão significativa da capacidade da linha.
- Alterações no design.
- Alterações na embalagem do produto.
- Transferência de um processo para outra instalação.
- Alterações no processo de submissão.
Para saber mais sobre os protocolos de validação e a sua importância no campo do fabrico de dispositivos médicos, fale connosco. Mantenha-se informado! Mantenha-se em conformidade!









