3 min de leitura
A rotulagem é uma parte integrante da comercialização de dispositivos médicos. O rótulo é uma peça de informação afixada ao dispositivo e/ou embalagem num formato legível por humanos. O principal objetivo da rotulagem é fornecer informações de segurança aos utilizadores, que podem ser profissionais de saúde, consumidores ou qualquer outra pessoa relevante.
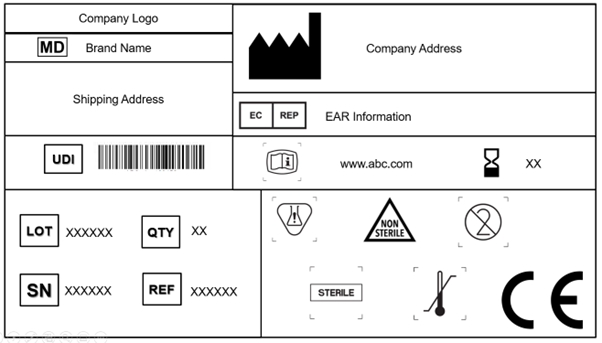
Todas as autoridades regulamentares globais têm certos requisitos de rotulagem. Da mesma forma, a UE detalhou os requisitos de rotulagem no Capítulo III, no Anexo I do Regulamento de Dispositivos Médicos da UE (EU MDR) 2017/745. O mais importante a notar é incluir todos os símbolos que abrangem as informações necessárias na rotulagem do dispositivo e nos documentos (folhetos, manuais, IFUs, etc.) que o acompanham.
Algumas das considerações críticas de rotulagem a ter em conta ao cumprir o EU MDR 2017/745 são-
1. Simbologia de Rotulagem de Dispositivos Médicos
Cada fabricante é obrigado a incorporar o símbolo de dispositivo médico, que indica que o produto fornecido ao mercado da UE é um dispositivo médico. É obrigatório afixar este símbolo no dispositivo e em todos os níveis de embalagem. Além disso, o rótulo deve apresentar o nome comercial e o nome original do dispositivo.
2. Dispositivos Especiais
Caso o produto seja um dispositivo especial ou personalizado, o seu estado deve ser mencionado na rotulagem. Por exemplo, se a finalidade do produto for apenas para investigação clínica, o rótulo deve indicá-lo explicitamente.
Para os dispositivos com materiais absorventes ou que se podem dispersar localmente no corpo humano, a rotulagem deve mencionar a composição do material e os detalhes quantitativos sobre os constituintes principais.
É exigida uma rotulagem explícita no caso de dispositivos de uso único e dispositivos estéreis. Para os dispositivos reprocessados, a rotulagem deve mencionar o número de vezes que podem ser reprocessados, o número de vezes que foram reprocessados até agora e o método de esterilização utilizado.
3. Presença de Substâncias Tóxicas
A declaração da presença de substâncias CMR (carcinogénicas, mutagénicas, tóxicas para a reprodução) e substâncias desreguladoras endócrinas é obrigatória nos rótulos se a concentração for superior a 0,1% p/p. A lista de tais substâncias deve ser afixada no dispositivo e/ou embalagem.
Além disso, um rótulo sobre a presença de derivados de sangue e tecidos (mesmo quando contidos na substância medicinal do dispositivo combinado) deve ser afixado nos dispositivos.
4. Normas Harmonizadas
O EU MDR 2017/745 reconhece e aceita a ISO 15223-1: 2021. O documento determina os símbolos a serem utilizados na Rotulagem de dispositivos médicos e nas suas embalagens. O Capítulo 3 (23.1,h) do Anexo I do EU MDR especifica que símbolos internacionalmente reconhecidos podem ser utilizados e, no caso de regiões onde estes símbolos não são reconhecidos, a descrição dos mesmos deve ser fornecida num documento juntamente com o dispositivo.
5. UDI
Os Artigos 27.º, 28.º, 29.º e o Anexo VI (A, B, C) detalham as regras e regulamentos para o UDI. O rótulo é agora obrigado a conter um suporte UDI [representação do UDI para Identificação Automática para Captura de Dados (AIDC) e Interpretação Legível por Humanos (HRI)] no dispositivo e também nos níveis de embalagem superiores. A embalagem superior do dispositivo (excluindo as embalagens de transporte) terá o seu próprio suporte UDI.
6. Informação Eletrónica para Utilização (eIFU)
O endereço web (URL) sob a forma de eIFUs também pode ser colocado na rotulagem do dispositivo médico, juntamente com as IFUs em papel. As eIFUs podem ser utilizadas no caso de dispositivos médicos implantáveis, implantáveis ativos, fixos e software (destinados também a leigos).
7. Informação dos Operadores Económicos (EOs)
O rótulo geralmente contém a informação do fabricante. No entanto, no caso de fabricantes estrangeiros, a informação do representante autorizado deve ser colocada nos rótulos comerciais.
8. Advertências e Precauções
Os avisos e precauções devem ser mencionados no rótulo do dispositivo. A informação sobre este aspeto pode ser mantida no mínimo, e os detalhes podem ser fornecidos nas IFU.
Os fabricantes também são obrigados a adaptar-se aos requisitos de rotulagem específicos de cada país. O requisito linguístico depende do Estado-Membro da UE. Pode ter um grande impacto na rotulagem, nas instruções de utilização (IFUs) e na embalagem do dispositivo em termos de tempo e custos.
Estes requisitos adicionais podem aumentar ainda mais o fardo do fabricante com a complexidade existente do processo de rotulagem. A falha neste aspeto pode tornar-se muito dispendiosa, envolvendo recolhas de produtos e etapas subsequentes para Ações Corretivas e Preventivas (CAPA).
Procura assistência com a rotulagem de acordo com o EU MDR? A Freyr oferece serviços abrangentes de rotulagem de dispositivos médicos. Sinta-se à vontade para contactar os nossos especialistas regulamentares agora em – sales@freyrsolutions.com









