
2 min de leitura
Uma 510(k) ou uma notificação pré-comercialização é uma submissão feita à United States Food and Drug Administration (US FDA) para demonstrar que o dispositivo a ser comercializado é seguro e eficaz, ou seja, substancialmente equivalente a um dispositivo legalmente comercializado ou a um dispositivo precedente. Os seguintes são os três (03) tipos de 510(k) que um fabricante de dispositivos médicos pode submeter:
- Tradicional
- Abreviado
- 510(k) Especial
Neste blog, examinaremos os casos em que a sua submissão se qualificaria para o segundo tipo, um 510(k) abreviado, de acordo com os requisitos da US FDA.
Um 510(k) abreviado é utilizado para demonstrar equivalência substancial a uma norma reconhecida, controlo especial ou orientação, utilizando uma Declaração de Conformidade (DoC). Numa submissão abreviada, os fabricantes demonstram equivalência substancial a normas reconhecidas com base na utilização de documentos de orientação ou DoCs, em vez de um dispositivo precedente, para facilitar a revisão da US FDA. Abaixo está o fluxograma para determinar a equivalência substancial para uma submissão 510(k) abreviada.
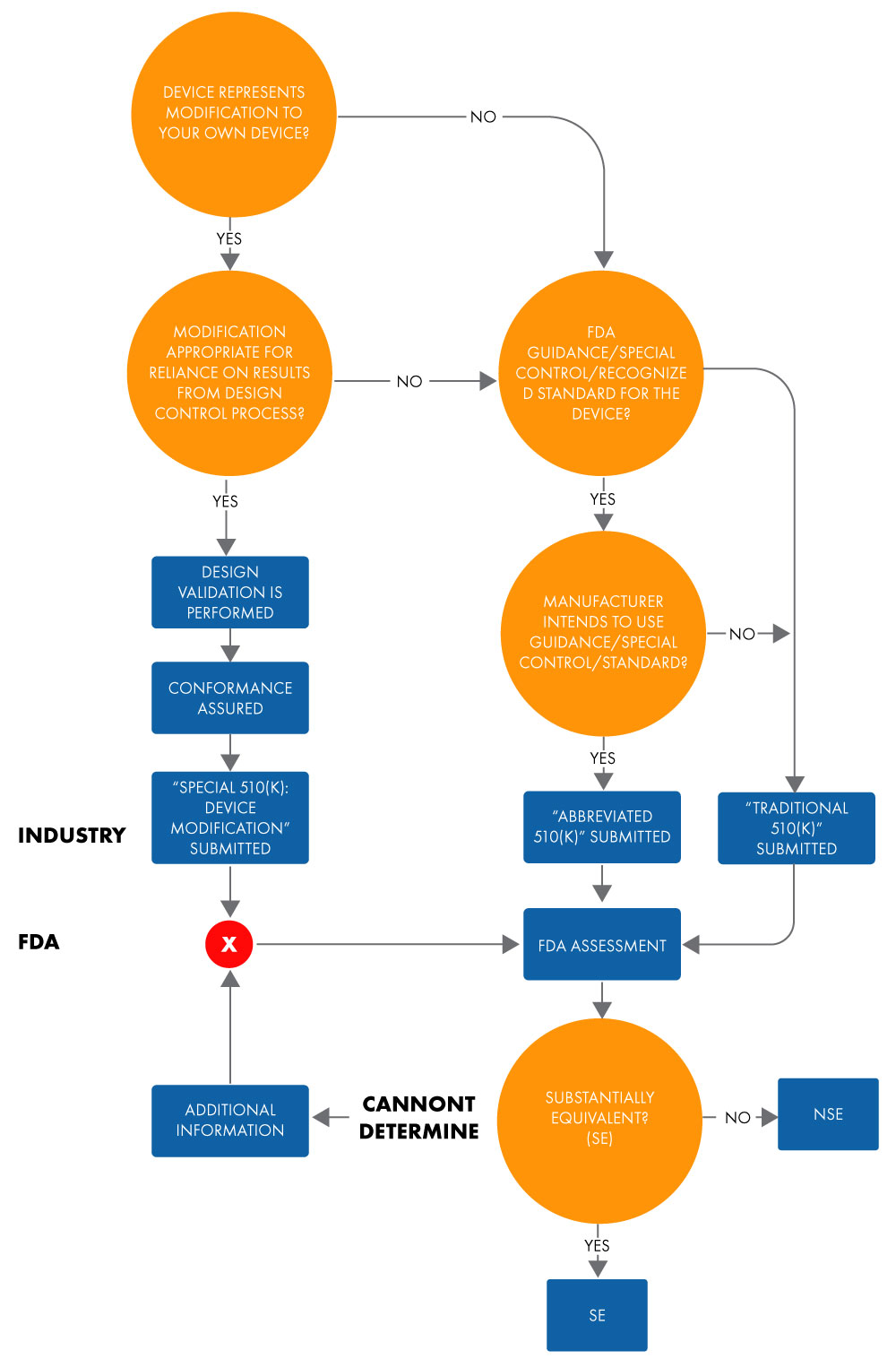
Figura 1: Intenção de Colocação no Mercado de Dispositivo Através de 510(k)
O termo “abreviado” sugere que este tipo de processo de aprovação 510(k) é mais curto. No entanto, isto não é totalmente verdade. Demora tanto tempo quanto uma aprovação 510(k) tradicional. O mesmo se aplica à documentação e aos custos. Além disso, o formato para ambos os 510(k) tradicionais e abreviados, em termos de divisão por capítulos e estrutura, é semelhante.
Ao submeter um 510(k) abreviado, deve basear-se nos elementos identificados em 21 CFR 807.87 (submissões 510[k] tradicionais). Pode optar por submeter um 510(k) abreviado quando a submissão se baseia no seguinte:
- Documento(s) de Orientação da FDA: Ao submeter um 510(k) abreviado, deve incluir um relatório de resumo que descreva a adesão ao documento de orientação relevante e como foi utilizado durante o desenvolvimento e teste do dispositivo.
- Demonstração de Conformidade com Controlos Especiais para o Tipo de Dispositivo: Deve cumprir os controlos especiais, tais como padrões de desempenho, Vigilância Pós-Comercialização (PMS), registos de pacientes, desenvolvimento e disseminação de diretrizes, recomendações, etc., que fornecem uma garantia razoável da segurança e eficácia do dispositivo. Uma submissão 510(k) abreviada que se baseia num(s) controlo(s) especial(is) deve incluir o seguinte: um relatório de resumo que descreve a adesão aos controlos especiais e como foram utilizados durante o desenvolvimento e teste do dispositivo.
- Como os controlos especiais foram utilizados para abordar um risco específico ou uma questão.
- Informações que descrevem quaisquer desvios dos controlos específicos e as tentativas do fabricante para os cumprir.
- Norma(s) de Consenso Voluntário: É necessário fornecer uma DoC para a norma reconhecida para uma submissão 510(k) abreviada que se baseia nela. Uma DoC deve incluir o seguinte:
- O nome e o endereço do requerente/patrocinador responsável pela DoC.
- Detalhes da identificação do produto/dispositivo, incluindo códigos de produto, nome comercial do dispositivo, número do modelo e quaisquer outros dados de identificação de produto únicos específicos da DoC em questão.
- Uma declaração de conformidade.
- Uma lista de normas às quais a DoC é aplicável, incluindo a(s) opção(ões) selecionada(s) para cada norma, se houver.
- O número de reconhecimento da FDA dos US para cada norma.
- A data e o local de emissão da DoC.
- A assinatura, o nome impresso e a função do patrocinador responsável pela DoC.
- Qualquer limitação(ões) à validade da DoC (por exemplo, por quanto tempo a declaração é válida, o que foi testado, concessões feitas sobre os resultados dos testes, etc.)
Em conclusão, um 510(k) abreviado é uma forma útil para os fabricantes de dispositivos demonstrarem equivalência substancial a normas reconhecidas ou controlos especiais, utilizando uma DoC. Para se qualificarem para um 510(k) abreviado, os fabricantes de dispositivos devem fornecer um relatório de resumo que explique a sua adesão aos documentos de orientação relevantes, demonstrar conformidade com os controlos especiais e fornecer DoCs para as normas reconhecidas. É, no entanto, importante notar que o processo de aprovação, a documentação e o custo de um 510(k) abreviado são semelhantes aos de um 510(k) tradicional.
O seu dispositivo médico qualifica-se para uma submissão 510(k) abreviada? Para qualquer assistência na apresentação da sua submissão 510(k) abreviada, contacte o nosso especialista regulamentar. Mantenha-se informado! Mantenha-se em conformidade!









